








Transition Metals (d-Block elements):
Elements formed by the filling of 3d, 4d and 5d shells of electrons comprise the d-block elements. They are also called transition elements because their position in the periodic table is between the s-block and p-block elements. Their properties are transitional between the highly reactive metallic elements of the s-block, which typically form ionic compounds, and the elements of the p-block, which are largely covalent.
Those elements or ions which have partly filled d sub-shell are called d-block elements or transition elements.
or
These elements in which the differentiating electrons occupy (n-1) d sub-shell, are d-block elements. They are called transition elements since, they show a transition in their properties from the left side (s-block) elements to the light side (p- block elements).
There are four d-series each starting with (n-1)d1ns2 constituting group3 to group 12 (or group IIIB to II B).
In the d-block, electrons are added to the penultimate shell, expanding it from 8 to 18 electrons. Typically the transition elements have an incompletely filled d level. Group 12 (the zinc group) has a d10 configuration and since the d shell is complete, compounds of these elements are not typical and show some differences from the others. The elements make up three complete rows of ten elements and an incomplete fourth row. The position of the incomplete fourth series is discussed with the f-block elements.
These elements include precious metals like silver gold, platinum and industrially important like iron, copper, nickel etc.
Electronic Configuration:
|
IIIB |
IVB |
VB |
VIB |
VIIB |
VIII |
VIII |
VIII |
IB |
IIB |
|
Sc21 Scandium |
Ti22 Zirconium |
V23 Vanadium |
Cr24 Chromium |
Mn25 Manganese |
Fe26 Iron |
CO27 Cobalt |
Mi28 Nickel |
Cu29 Copper |
Zn30 Zinc |
|
Y39 Yttrium |
Zr40 Titanium |
Nb41 Niobium |
Mo42 Molybetenium |
Te43 Technelium |
Ru44 Ruthenium |
Rh45 Rhodium |
Pd46 Palladium |
Ag47 Silver |
Cd48 Cadmium |
|
La57 Lanthanum |
Hf72 Hafnium |
Ta73 Tantalum |
W74 Tungsten |
Re75 Rhenium |
Ds76 Osmium |
Ir77 Iridium |
Pt78 Platinum |
Au79 Gold |
Hg80 Mercury |
|
Ac89 Actinium |
Rf104 Rutherford |
Db105 Dubnium |
Sg106 Seabagium |
Bh107 Bohrium |
Hs108 Hassium |
Mt109 Meitherium |
Uun110 Un-un-unium |
Uun111 Un-un-un-ium |
Unb112 Un-ub-bium |
Electronic configuration:
1st Series
|
[Ar] |
Sc |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu |
Zn |
|
4s |
2 |
2 |
2 |
1 |
2 |
2 |
2 |
2 |
1 |
2 |
|
3d |
1 |
2 |
3 |
5 |
5 |
6 |
7 |
8 |
10 |
10 |
2nd Series
|
[Ar] |
Sc |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu |
Zn |
|
4s |
2 |
2 |
2 |
1 |
2 |
2 |
2 |
2 |
1 |
2 |
|
3d |
1 |
2 |
3 |
5 |
5 |
6 |
7 |
8 |
10 |
10 |
3rd Series
|
[Xe] |
La |
Hf |
Ta |
W |
Re |
Os |
Ir |
Pt |
Au |
Hg |
|
6s |
2 |
2 |
2 |
2 |
2 |
2 |
2 |
1 |
1 |
2 |
|
5d |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
9 |
10 |
10 |
Strictly speaking in Zn, Cd & Hg the differentiating electron enters the ns subshell & not the (n-1)d subshell hence, according to definition, they should be excluded from d-block elements. But, since they have almost all of their other properties similar to those of d-block elements, they are placed with them.
The general electronic configuration of d block is
[Noble gas] ns1–2 (n-1)d1–10 [Only Pd has 5s0 4d10]
Filling of the electron follows the sequence of increasing order of energy as given by Aufbau principle, alternatively by (n +λ) rule, which states that electron are filled in ns earlier than (n-1) d so as 4s is filled before 3d
5s before 4d and 6s before 5d so on ……..
But this sequence is disturbed in between, when d-orbital is about to be half or fully filled. e.g., at Cr & Cu in 1st series, Nb to Ag in 2nd & Pt & Au in 3rd series.
Cr 24 = 4s1 3d5
Cu29 = 4s1 3d10
Mo42 = 5s1 4d5
Pd46 = 5s0 4d10
Ag47 = 5s1 4d10
Pt78 = 6s1 5d9 or 6s0 5d10
Au79 = 6s1 5d10
The above elements of d-block have a similar tendency to fill their d-subshell either partially or completely and while doing so, they violate the Aufbau law. This is because half filled or completely filled d-levels are more stable due to greater exchange energy.
This exchange of e– between ns & (n-1) d is possible with constraints of stability only due to small energy difference between these subshells.
It should be noted that when e– are to be removed from 3d elements, they are removed from the outermost shell i.e., ns and not the penultimate i.e., (n-1)d so.
Illustration 1: Though copper, silver and gold have completely filled sets of d-orbitals yet they are considered as transition metals. Why?
Solution: These metals in their common oxidation states have incompletely filled d-orbitals, e.g., Cu2+ has 3d9 and Au3+ has 5d8 configuration.
Illustration 2: Which out of the following is/are transition element/s and why?
Solution: Ag and Au are transition elements because they have incompletely filled d-subshell.
Abundance:
Three of the transition metals are very abundant in the earth’s crust. Fe – the fourth most abundant element by weight, Ti – the ninth and Mn – the twelfth. The first row of transition elements largely follow Harkins’ rule that elements with an even atomic number are in general more abundant than their neighbours with odd atomic numbers. Manganese is an exception. The second and third row elements are much less abundant than the first row. Tc does not occur in nature. Of the last six elements in the second and third rows (Tc. Ru. Rh. Pd, Ag. Cd Re, Os. lr, Pt, Au, Hg) none occurs to an extent of more than 0.16 parts per million (ppm) in the earth’s crust.
Abundance (in ppm):
|
Sc |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu |
Zn |
|
25 |
6320 |
136 |
122 |
1060 |
60000 |
29 |
99 |
68 |
76 |
|
Y |
Zr |
Nb |
Mo |
Tc |
Ru |
Rh |
Pd |
Ag |
Cd |
|
31 |
162 |
20 |
1.2 |
– |
0.0001 |
0.0001 |
0.015 |
0.08 |
0.16 |
|
La |
Hf |
Ta |
W |
Re |
Os |
Ir |
Pt |
Au |
Hg |
|
35 |
2.8 |
1.7 |
1.2 |
0.0007 |
0.005 |
0.001 |
0.01 |
0.004 |
0.08 |
Physical properties:
1) Metallic character:
In the d-block elements the penultimate shell of electrons is expanding. Thus they have many physical and chemical properties in common. Thus all the transition elements are metals. They are therefore good conductors of electricity and heat, have a metallic lustre and are hard, strong and ductile. They also form alloys with other metals.
All d-block elements are metals. Unlike s-blocks they are hard, malleable and ductile with very high mp & bp’s. Hardness and high mp indicate strong binding forces in their crystals.
They are good conductor of electricity and have metallic lusture due to delocalization e– over the entire crystal structure.
In a transition series the no. of unpaired e– increases from IIIB to VI B and then decreases (due to pairing up). So, the metallic lattice becomes stronger upto group VIB whereafter it decreases slowly.
Moving across a period, there is gradual decrease in the electropositive character. The strong metallic bonding in transition metals is due to greater effective nuclear charge and large no. of valence electrons (here all the electrons outside the noble gas configuration [i.e., ns & (n-1)d] are considered to be valence e–)
It is only the metallic bonding which accounts the metals to be good conductor of heat and electricity and their high density.
Illustration3: Transition metals exhibit higher enthalpies of atomization. Explain why?
Solution: Enthalpy of atomsation is the amount of heat required to break the metal lattice to get free atoms. As transition metals contain a large number of unpaired electrons, they have strong inter atomic attractions (metallic bonds). Hence, they have high enthalpies of atomization.
Atomic and Ionic Radii:
The covalent radii of the elements (see table) decrease from left to right across a row in the transition series, until near the end when the size increases slightly. On passing from left the right, extra protons are placed in the nucleus and extra orbital electrons are added. The orbital electrons shield the nuclear charge incompletely (d electrons shield less efficiently than p electrons, which in turn shield less effectively than s electrons). Because of this poor screening by d electrons, the nuclear charge attracts all of the electrons more strongly: hence a contraction in size occurs.
Atoms of the transition elements are smaller than those of the Group 1 or 2 elements in the same horizontal period. This is partly because of the usual contraction in size across a horizontal period discussed above, and partly because the orbital electrons are added to the penultimate d shell rather than to the outer shell of the atom. Interposed between lanthanum and hafnium are the 14 lanthanide elements, in which the antepenultimate 4f shell of electrons is filled.
Table: Covalent radii of the transition elements (Å)
|
K |
Ca |
Sc |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu |
Zn |
|
1.57 |
1.74 |
1.44 |
1.32 |
1.22 |
1.17 |
1.17 |
1.17 |
1.16 |
1.15 |
1.17 |
1.25 |
|
Rb |
Sr |
Y |
Zr |
Nb |
Mo |
Tc |
Ru |
Rh |
Pd |
Ag |
Cd |
|
2.16 |
1.91 |
1.92 |
1.45 |
1.34 |
1.29 |
|
1.24 |
1.25 |
1.28 |
1.34 |
1.41 |
|
Cs |
Ba |
La * |
Hf |
Ta |
W |
Re |
Os |
Ir |
Pt |
Au |
Hg |
|
2.35 |
1.98 |
1.69 |
1.44 |
1.34 |
1.30 |
1.28 |
1.26 |
1.26 |
1.29 |
1.34 |
1.44 |
*14 Lanthanide elements
Table: The effect of the lanthanide contraction on ionic radii
|
Ca2+ |
1.00 |
Sc3+ |
0.745 |
|
Ti4+ |
0.605 |
V3+ |
0.64 |
|
Sr2+ |
1.18 |
Y3+ |
0.90 |
|
Zr4+ |
0.72 |
Vb3+ |
0.72 |
|
Ba2+ |
1.35 |
La3+ |
1.032 |
* |
Hf4+ |
0.71 |
Ta3+ |
0.72 |
*Lanthanides
There is a gradual decrease in size of the 14 lanthanide elements from cerium to lutetium. This is called the lanthanide contraction. The lanthanide contraction cancels almost exactly the normal size increase on descending a group of transition elements. The covalent radius of I-If and the ionic radius of Hf are actually smaller than the corresponding values for Zr. The covalent and ionic radii of Nb are the same as the values for Ta. Therefore the second and third row transition elements have similar radii. As a result they also have similar lattice energies, solvation energies and ionization energies. Thus the differences in properties between the first row and second row elements are much greater than the differences between the second and third row elements. The effects of the lanthanide contraction are less pronounced towards the right of the d-block. However, the effect still shows to a lesser degree in the p-block elements which follow.
Illustration 4: Which out of the two, La(OH)3 and Lu(OH)3, is more basic and why?
Solution: La (OH)3 is more basic than Lu (OH)3. As the size of the lanthanide ions decreases from La3+ to Lu3+, the covalent character of the hydroxides increases (Fajan’s rules). Hence, the basic strength decreases from La (OH)3 to Lu (OH)3.
Density:
The atomic volumes of the transition elements are low compared with elements in neighbouring Groups 1 and 2. For a transition series density increases across the period on moving left to right, attains a maximum value at group VIII & then decreases. However moving down a group density increases substantially since atomic radii remain almost same, while mass is increasing (almost doubled). The reason is the smaller radii and close packed structure. In addition, the extra electrons added occupy inner orbitals. Consequently the densities of the transition metals are high. Practically all have a density greater than 5 g cm–3 (The only exceptions are Sc 3.0 g cm–3 and Y and Ti 4.5 g cm–3. The densities of the second row are high and third row values are even higher. The two elements with the highest densities are osmium 22.57g cm–3 and iridium 22.61 g cm–3. So, a football made of osmium or iridium measuring 30 cm in diameter would weigh 320 kg or almost one third of a tonne!
|
Transition series |
Element |
At radius (Å) |
At mass (amu) |
Density (g cm–3) |
Element |
At radius (Å) |
At mass (amu) |
Density (g cm–3) |
|
IInd series |
Nb |
1.46 |
93 |
8.4 |
M0 |
1.39 |
96 |
10.4 |
|
IIIrd series |
Ta |
1.46 |
181 |
16.6 |
W |
1.36 |
184 |
19.3 |
Note: Osmium (Os) has the highest density of 22.6 g cm –3 of all the elements.
MP & BP: The melting and boiling points of the transition elements are generally very high. Transition elements typically melt above 1000°C. Three elements melt above 3000°C (Ta 3000°C, W 3410°C and Re 3180°C). There are a few exceptions. The melting points of La and Ag are just under 1000°C (920°C and 961°C respectively). Other notable exceptions are Zn (420°C), Cd (321 °C) and Hg which is liquid at room temperature and melts at —38°C. The last three behave atypically because the d shell is complete, and d electrons do not participate in metallic bonding. The high melting points are in marked contrast to the low melting points for the s-block metals Li (181 °C) and Cs (29°C).
Ionization energies:
There is an increase in the ionization energy but the increase is less compared to s or p block elements of the same period. For example: IE of Sc, Ti, V & Cr are fairly close to one another. Similarly values for Fe, Co, Ni & Cu are also very close. The reason being the screening effect of d-electrons which compensates the increment in nuclear charge. However the values lie between s & p blocks on either side.
The change for 2nd IE is more smooth with only variation at Cr (3d5) and Cu (3d10) due to most stable d subshells – configurations. Similarly for 3rd IE Mn & Zn have sufficiently higher values for the same reason.
Reactivity:
Many of the metals are sufficiently electropositive to react with mineral acids, liberating H2. A few have low standard electrode potentials and remain unreactive or noble. Noble character is favoured by high enthalpies of sublimation, high ionization energies and low enthalpies of solvation. (Born-Haber cycle) The high melting points indicate high heats of sublimation. The smaller atoms have higher ionization energies. But this is offset by small ions having high solvation energies. This tendency to noble character is most pronounced for the platinum metals (Ru, Rh, Pd, Os, Ir, Pt) and gold.
Oxidation state:
One of the most striking features of the transition elements is that the elements usually exist in several oxidation states. Furthermore, the oxidation states change in units of one, e.g. Fe (II) and Fe (III); Cu (I) and Cu (II). The oxidation states shown by the transition elements may be related to their electronic structures. Calcium, the s-block element preceding the first row of transition elements, has the electronic structure:
Ca = 1s22s22p63s23p6 4s2
It might be expected that the next ten transition elements would have this electronic arrangement with from one to ten d electrons added in a regular way: 3d1, 3d2, 3d3 …… 3d10. This is true except in the cases of Cr and Cu. In these two cases one of the s electrons moves into the d shell, the additional stability when the d orbitals are exactly half filled or completely filled.
In contrast to s-block elements which have +I & +II respectively, transition elements can exist in a variety of ox. states. These states depend on the no. of e– in outermost subshell ‘plus’ penultimate subshell. The maximum valency that an element of d-block can exhibit is found to increase from 3rd group to 7th or 8th thereafter it decreases upto 12th.
Oxidation states of d-block elements:
|
Element |
Sc |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Mi |
Cu |
Zn |
|
Oxidation states |
(+2) |
+2 |
+2 |
+1 |
+2 |
+2 |
+2 |
+2 |
+2 |
+2 |
|
|
+3 |
+3 |
+3 |
+3 |
+3 |
+3 |
+3 |
+3 |
|
|
|
|
|
+4 |
+4 |
+4 |
+4 |
+4 |
+4 |
|
|
|
|
|
|
+5 |
|
|
|
|
|
|
|
|
|
|
|
|
+6 |
+6 |
+6 |
|
|
|
|
|
|
|
|
|
|
+7 |
|
|
|
|
|
|
Note the difference between variable valency of p block (heavy atoms) with d blocks – in p-block the difference of the two states was always 2 i.e., TlI & TlII PbII & PbIV, SbIII & SbIV etc due to inert inner 2s e– but here the difference is one i.e., FeII, FeIII, CoII & CoIII, CuI & CuII. Here the reason is mainly the no. of e– in d subshell.
Thus Sc could have an oxidation number of (+ II) if both s electrons are used for bonding and (+III) when two s and one d electrons are involved. Ti has an oxidation state (+II) when both s electrons are used for bonding, (+III) when two s and one d electrons are used and (+IV) when two sand two d electrons are used. Similarly, V shows oxidation numbers (+II), (+III) (+IV) and (+VI). In the case of Cr, by using the singles electron for bonding, we get an oxidation number of (+I): hence by using varying numbers of d-electrons oxidation states of (+II), (+III), (+V) and (+Vl) are possible. Mn has oxidation states (+II). (+III), (+IV), (+V), (+V1) and (+VII). Among these first five elements, the correlation between electronic structure and minimum and maximum oxidation states in simple compounds is complete. In the highest oxidation state of these first five elements, all of the s and d electrons are being used for bonding. Thus the properties depend only on the size and valency and consequently show some similarities with elements of the main groups in similar oxidation states. For example, SO42– (Group 16) and CrO42– (Group 6) are isostructural as are SiCl4 (Group 14) and TiCl4 (Group 4). Once the d configuration is exceeded, i.e. in the last five elements the tendency for all the d electrons to participate in bonding decreases. Thus Fe has a maximum oxidation state of (+VI). However, the second and third elements in this group attain a maximum oxidation state of (+ VIII), in RuO4 and OsO4. This difference between Fe and the other two elements Ru and Os is attributed to the increased size of the latter two elements where all the 6d and 2s e– be removed.
These facts may be conveniently memorized, because the oxidation states form a regular ‘pyramid’ as shown in Table. Only Sc(+II) and Co(+V) are in doubt. The oxidation number of all elements in the elemental state is zero. In addition, several of the elements have zero valent and other low-valent states in complexes. Low oxidation states occur particularly with n bonding ligands such as carbon monoxide and dipyridyl. Similar but not identical pyramids of oxidation states are found in the second and third rows of transition elements. The main differences are as follows:
1. In Group 8 (the iron group) the second and third row elements show a maximum oxidation state of (+V1Jl) compared with (+VI) for Fe.
2. The electronic structures of the atoms in the second and third rows do not always follow the pattern of the first row. The structures of group 10 elements (the nickel group) are:
Ni 3d8 4s2
Pd 4d10 5s0
Pt 5d9 6s1
Since a full shell of electrons is a stable arrangement, the place where this occurs is of importance.
The 4 levels are complete at copper, palladium and gold in their respective series.
|
Ni |
3d8 |
4s2 |
Cu |
3d10 |
4s1 |
Zn |
3d10 |
4s2 |
|
Pd |
4d10 |
5s0 |
Ag |
4d10 |
5s1 |
Cd |
4d10 |
5s2 |
|
Pt |
5d9 |
6s1 |
Au |
5d10 |
6s1 |
Hg |
5d10 |
6s2 |
The maximum ox. st of an element with reasonable stability corresponds to the value of no. of e– in ns + (n-1)d subshells upto Mn; thereafter it decreases upto Cu. (s, p, d & f should not be called orbitals. They actually are subshells; they are made up of orbitals e.g., p subshell comprises of px, py & pz orbitals).
There is a difference in valency & ox. state. Oxidation no. is the residual charge left over an atom of a compound, if all the rest atoms are replaced in ionic form, while valency is the no. of e– that take bart in formation of a compound – either through ionic or covalent bond formation. Abrupt decrease of stability in the higher stable oxidation state is observed. Due to no. of e– in ns subshell the minimum ox no. I or II can be accounted for, but there is no regular rule for maximum ox. st.
Stability of the various oxidation states:
Compounds are regarded as stable if they exist at room temperature, are not oxidized by the air, are not hydrolysed by water vapour and do not disproportionate or decompose at normal temperatures. Within each of the transition Groups 3—12, there is a difference in stability of the various oxidation states that exist. In general the second and third row elements exhibit higher coordination numbers, and their higher oxidation states are more stable than the corresponding first row elements. This can be seen from Table. This gives the known oxides and halides of the first, second and third row transition elements. Stable oxidation states form oxides, fluorides, chlorides, bromides and iodides. Strongly reducing states probably do not form fluorides and/or oxides, but may well form the heavier halides. Conversely, strongly oxidizing states form oxides and fluorides, but not iodides.
Oxides and halides of the first row
|
|
|
Sc |
Ti |
V |
Cr |
Mn |
Fe |
Co |
Ni |
Cu |
Zn |
|
+ II |
O |
|
TrO |
VO |
CrO |
MnO |
FeO |
CeO |
NiO |
CuO |
ZnO |
|
|
F |
|
|
VF2 |
CrF2 |
MnF2 |
FeF2 |
CoF2 |
NiF2 |
CuF2 |
ZNF2 |
|
|
Cl |
|
HCl2 |
VCl2 |
CrCl2 |
MnCl2 |
FeCl2 |
CoCl2 |
NiCl2 |
CuCl2 |
ZnCl2 |
|
|
Br |
|
TiBr2 |
VBr2 |
CrBr2 |
MnBr2 |
FeBr2 |
CoBr2 |
NiBr2 |
CuBr2 |
ZnBr2 |
|
|
I |
|
Til2 |
Vl2 |
Crl2 |
Mnl2 |
Fel2 |
Col2 |
Nil2 |
|
Znl2 |
|
+III |
O |
Se2O3 |
Ti2O3 |
V2O3 |
Cr2O3 |
Mn2O3 |
(Co2O3)b |
(Ni2O3)b |
|
|
|
|
|
F |
SeF3 |
TiF3 |
CrF3 |
MnF3 |
FeF3 |
CoF3 |
|
|
|
|
|
|
Cl |
SeCl3 |
TiCl3 |
VCl3 |
|
FeCl3 |
|
FeCl3 |
|
|
|
|
|
Br |
SeBr3 |
TiBr3 |
VBr3 |
CrBr3 |
|
|
FeBr3 |
|
|
|
|
|
I |
Sel3 |
Til3 |
Til3 |
Vl3 |
Crl3 |
|
|
|
|
|
|
+IV |
O |
TiO2 |
VO2 |
CrO2 |
MnO2 |
|
(CoO2)b |
N2O3b |
|
|
|
|
|
F |
|
TiF4 |
VF4 |
CrF4 |
MnF4 |
|
|
|
|
|
|
|
Cl |
|
TiCl4 |
VCl4 |
CrCl4 |
|
|
|
|
|
|
|
|
Br |
TiBr4 |
VBr4 |
CrBr4g |
|
|
|
|
|
|
|
|
|
I |
Til4 |
|
Crl4 |
|
|
|
|
|
|
|
|
+V |
O |
|
V2O3 |
|
|
|
|
|
|
|
|
|
|
F |
|
VF4 |
CrF3 |
|
|
|
|
|
|
|
|
|
Cl |
|
|
|
|
|
|
|
|
|
|
|
|
Br |
|
|
|
|
|
|
|
|
|
|
|
|
I |
|
|
|
|
|
|
|
|
|
|
|
+VI |
O |
|
|
CrO4 |
|
|
|
|
|
|
|
|
|
F |
|
|
(CrF0) |
|
|
|
|
|
|
|
|
|
Cl |
|
|
|
|
|
|
|
|
|
|
|
|
Br |
|
|
|
|
|
|
|
|
|
|
|
|
I |
|
|
|
|
|
|
|
|
|
|
|
+VII |
O |
|
|
|
|
Mn2O7 |
|
|
|
|
|
|
|
F |
|
|
|
|
|
|
|
|
|
|
|
|
Cl |
|
|
|
|
|
|
|
|
|
|
|
|
Br |
|
|
|
|
|
|
|
|
|
|
|
|
I |
|
|
|
|
|
|
|
|
|
|
|
Other compounds |
|
|
|
|
Mn2O3 |
Fe3O4 |
Co3O4 |
|
Cu2O |
|
|
|
|
|
|
|
|
|
|
|
|
CuCl |
|
|
|
|
|
|
|
|
|
|
|
|
CuBr |
|
|
|
|
|
|
|
|
|
|
|
|
Cul |
|
|
For the first transition series the most common ox. st. with all the elements is +2 which is the minimum ox. st. shown by the d block elements (except group 11 or IB and Hg which exhibit +1 in addition to higher values).
Some transition metals show zero oxidation state with strong p acid ligands*, and sometimes even ‘negative’ values are observed; e.g.,
Ni (CO)4 Ni (0)
Fe(CO)5 Fe(0)
Na (Mn (CO)5] Mn (-I)
The compounds wherein elements reflect their highest oxidation state are those of having small sized & high electronegativity atoms as anions (like F– & O2–)
The maximum ox. st in
|
1st transition series |
Mn |
(VII) |
(3d5 4s2) |
|
2nd transition series |
Ru |
(VIII) |
4d6 5s2) |
|
3rd transition series |
Os |
(VIII) |
(5d66s2) |
Illustration 5: Why do transition elements show variable oxidation states?
Solution: In the transition elements, the energies of (n – 1) d –orbitals and ns orbitals are very close. Hence, electrons from both can participate in bonding.
Illustration 6: Decide giving reason which one of the following pairs exhibits the property indicated:
V or Mn exhibits more number of oxidation states (Atomic numbers: Sc = 21, Cr = 24, V = 23, Mn = 25)
Solution: Mn exhibits more number of oxidation states.
π-acid ligands:
These are the ions or molecules which have π bond in them and are able to form back bonding with metal due to presence of a vacant orbital. These vacant orbitals of the ligand can accept electron pair from metal & thus behaving as a ligand (donor) as well as an acid (a Lewis acid – an acceptor).
As usually ligands are the electrons donors and they donate a lone pair to metal and behave as a Lewis base. But in case of CO, i.e., carbon monoxide due to resonating structure has an extra lone pair which is donated to the metal atom forming an organometallic (actually C → M) bond.
At the same time, the increased charge density over the metal atom is localized over the available antibonding orbitals of CO forming a π-bond with the filled d-orbital of metal.
Tendency to form complexes:
Since these elements have either empty d-orbitals or they can achieve a configuration with it which can accommodate e– from the ligands (the molecules with at least an unshared e– paired like, CO, NO, NH3, H2O, F–, Cl– CN– etc).
The factor that determine the extent of complex formation are
(i) Small size and high charge density
(ii) Presence of vacant d orbitals
Because of presence of coordinate bond between metal cation and ligands, the complexes are called coordination compounds.
Complexes:
The transition elements have an unparalleled tendency to form coordination compounds with Lewis bases, that is with groups which are able to donate an electron pair. These groups are called ligands. A ligand may be a neutral molecule such as NH3 or an ion such as Cl– or CN–. Cobalt forms more complexes than any other element, and forms more compounds than any other element except carbon.
Co3+ + 6NH3 → [Co(NH3)6]3+
Fe2+ + 6CN– → [Fe(CN)6]4–
This ability to form complexes is in marked contrast to the s- and p-block elements which form only a few complexes. The reason why transition elements are so good at forming complexes is that they have small, highly charged ions and have vacant low energy orbitals to accept lone pairs of electrons donated by other groups or ligands. Complexes where the metal is in the (+III) oxidation state are generally more stable than those where the metal is in the (+II) state.
Some metal ions form their most stable complexes with ligands in which the donor atoms are N, O or F. Such metal ions include Group 1 and 2 elements, the first half of the transition elements, the lanthanides and actinides, and the p-block elements except for their heaviest member. These metals are called class-a acceptors, and correspond to ‘hard’ acids (see ‘Acids and bases’). In contrast the metals Rh, Ir, Pd, Pt, Ag, Au and Hg form their most stable complexes with the heavier elements of Groups 15, 16 and 17. These metals are called class-b acceptors, and correspond to ‘soft’ acids. The rest of the transition metals, and the heaviest elements in the p-block, form complexes with both types of donors, and are thus ‘intermediate’ in nature. These are shown (a/b) in Table.
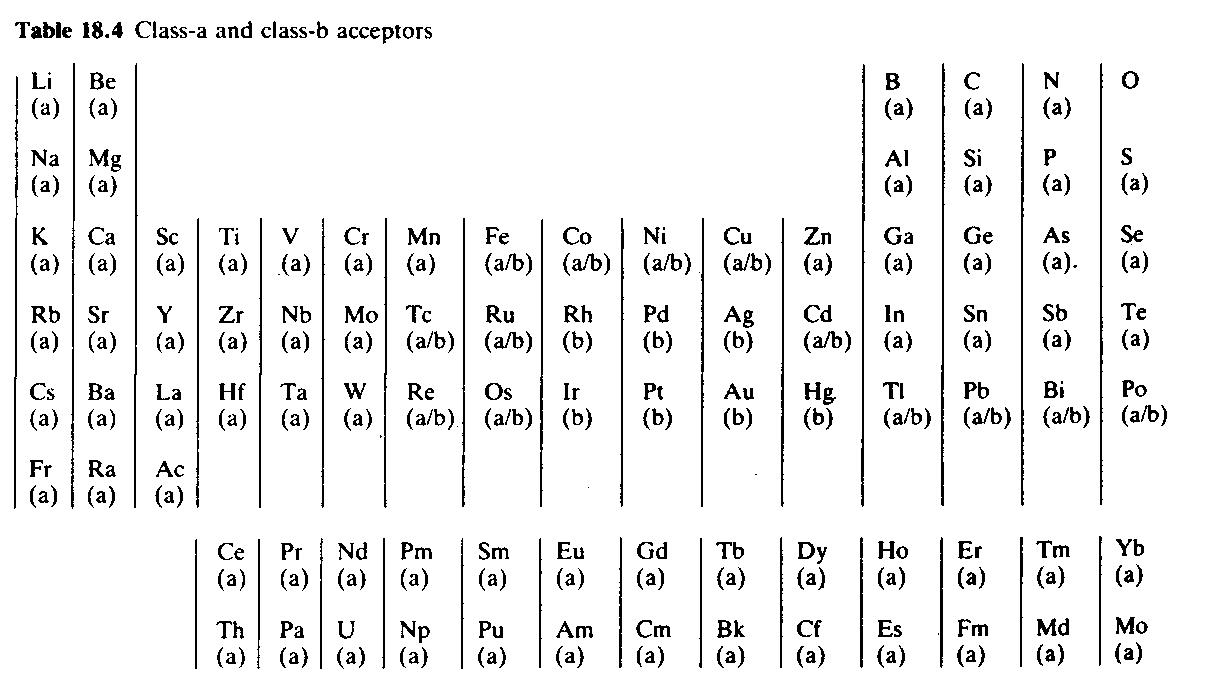
The nature of coordination complexes and the important crystal field theory of bonding are discussed in the next chapter.
Standard electrode potential (E0) and chemical reactivity:
The standard Red. Pot. (E0 Mn+/M) of the d block elements are ‘negative’ (i.e., less than that of H, 0.00 volt) except Cu, Ag, Au, Pt, Hg but ‘less negative’ than s block elements (Na, K, Ca, Mg). So they are comparatively less reactive and their reducing prop. is less than alkali & alkaline earth metals.
The less value of E0 is due to high ionization energy & sublimation energy (or enthalpy of atomization) coupled with less hydration enthalpy, as
Colour:
Many ionic and covalent compounds of transition elements are coloured. In contrast compounds of the s- and p-block elements are almost always white. When light passes through a material it is deprived of those wave lengths that are absorbed. If absorption occurs in the visible region of the spectrum, the transmitted light is coloured with the complementary colour to the colour of the light absorbed. Absorption in the visible and UV regions of the spectrum is caused by changes in electronic energy. Thus the spectra are sometimes called electronic spectra. It is always possible to promote an electron from one energy level to another. However, the energy jumps are usually so large that the absorption lies in the UV region. Special circumstances can make it possible to obtain small jumps in electronic energy which appear as absorption in the visible region.
Illustration 7: Why Zn2+ salts are white while Cu2+ salts are blue?
Solution: It is because of presence of unpaired electrons in d subshells. Cu2+ has incompletely filled d-orbitals (3d9).
Illustration 8: Scandium forms no coloured ions, yet it is regarded as a transition element. Explain why?
Solution: Scandium in the ground state has one electron in the 3d-subshell, it is regarded as a transition element.
Polarization:
NaCI, NaBr and NaI are all ionic, and are all colourless. AgCI is also colourless. Thus the halide ions CI–, Br– and I–, and the metal ions Na+ and Ag+ are typically colourless. However, AgBr is pale yellow and AgI is yellow. The colour arises because the Ag ion polarizes the halide ions. This means that it distorts the electron cloud, and implies a greater covalent contribution. The polarizability of ions increases with size: thus I– is the most polarized, and is the most coloured. For the same reason Ag2CO3 and Ag3PO4 are yellow, and Ag2O and Ag2s are black.
Incompletely filled d or f shells:
Colour may arise from an entirely different cause in ions with incomplete d or f shells. This source of colour is very important in most of the transition metal ions. In a free isolated gaseous ion the five d orbitals are degenerate that is they are identical in energy. In real life situations the ion will be surrounded by solvent molecules if it is in solution, by other ligands if it is in a complex. or by other ions if it is in a crystal lattice. The surrounding groups affect the energy of some d orbitals more than others.
Thus the d orbitals are no longer degenerate, and at their simplest they form two groups of orbitals of different energy. Thus in transition element ions with a partly filled d shell it is possible to promote electrons from one d level to another d level of higher energy. This corresponds to a fairly small energy difference, and so light is absorbed in the visible region. The colour of a transition metal complex is dependent on how big the energy difference is between the two d levels. This in turn depends on the nature of the ligand, and on the type of complex formed. Thus the octahedral complex [Ni(NH3)6]2+ is blue, [Ni(H2O)6]2+ is green and [Ni(NO2)6]4– is brown-red. The colour changes with the ligand used. The colour also depends on the number of ligands and the shape of the complex formed.
The source of colour in the lanthanides and the actinides is very similar, arising from f-f transitions. With the lanthanides the 4f orbitals are deeply embedded inside the atom, and are well shielded by the 5s and 5p electrons. The f electrons are practically unaffected by complex formation: hence the colour remains almost constant for a particular ion regardless of the ligand. The absorption bands are also very narrow.
Some compounds of the transition metals are white, for example ZnSO4 and TiO2 In these compounds it is not possible to promote electrons within the d level. Zn has a d10 configuration and the d level is full. Ti has a d° configuration and the d level is empty. In the series Sc(+III), Ti(+IV), V(+V), Cr(+VI) and Mn(+VII) ions may all be considered to have an empty d shell: hence d—d spectra are impossible and they should be colourless. However, as the oxidation number increases these states become increasingly covalent. Rather than form highly charged simple ions, oxoions are formed TiO2+ VO2+, VO42– CrO42– and MnO4–. VO2+ is pale yellow, but CrO42– is strongly yellow coloured, and MnO4– has an intense purple colour in solution though the solid is almost black.
The colour arises by charge transfer. In MnO4– an electron is momentarily transferred from O to the metal, thus momentarily changing O2– to O– and reducing the oxidation state of the metal from Mn(VII) to Mn(VI).
The s-and p-block elements do not have a partially filled d shell so there cannot be any d—d transitions. The energy to promote an s or p-electron to a higher energy level is much greater and needs ultraviolet light to be absorbed. Thus compounds of s- and p-block elements typically are not coloured.
Magnetic Properties:
When a substance is placed in a magnetic field of strength H. the intensity of the magnetic field in the substance may be greater than or less than H. If the field in the substance is greater than H, the substance is paramagnetic. It is easier for magnetic lines of force to travel through a paramagnetic material than through a vacuum. Thus paramagnetic materials attract lines of force, and, if it is free to move, a paramagnetic material will move from a weaker to a stronger part of the field. Paramagnetism arises as a result of unpaired electron spins in the atom.
If the field in the substance is less than H, the substance is diamagnetic. Diamagnetic materials tend to repel lines of force. It is harder for magnetic lines of force to travel through diamagnetic materials than through a vacuum, and such materials tend to move from a stronger to a weaker part of a magnetic field. in diamagnetic compounds all the electron spins are paired.
The paramagnetic effect is much larger than the diamagnetic effect.
It should be noted that Fe, Co and Ni are ferromagnetic. Ferromagnetic materials may be regarded as a special case of paramagnetism in which the moments on individual atoms become aligned and all of them point in the same direction. When this happens the magnetic susceptibility is greatly enhanced compared with if all the moments behaved independently. Alignment occurs when materials are magnetized, and Fe, Co and Ni can form permanent magnets. Ferromagnetism is found in several of the transition metals and their compounds.
It is also possible to get antiferromagnetism by pairing the moments on adjacent atoms which point in opposite directions. This gives a magnetic moment less than would be expected for an array of independent ions. It occurs in several simple salts of Fe, Mn and Gd Since ferromagnetism and antiferromagnetism depend on orientation, they disappear in solution.
Many compounds of the transition elements are paramagnetic, because they contain partially filled electron shells. If the magnetic moment is measured, the number of unpaired electrons can be calculated. The magnetochemistry of the transition elements shows whether the d electrons are paired. This is of great importance in distinguishing between high-spin and low-spin octahedral complexes.
There are two common methods of measuring magnetic susceptibilities: the Faraday and the Gouy methods.
The magnetic moment of a transition metal can give important information about the number of unpaired electrons present in the atom and the orbitals that are occupied, and sometimes indicates the structure of the molecule or complex. lithe agnetic moment is due entirely to the spin of unpaired electrons p. then
where S is the total spin quantum number. This gives the magnetic moment in SI units of JT’, and the magnetic moment in Bohr magnetons is given by (4S(S + I)). This equation is related to the number of unpaired electrons n by the equation:
The observed magnetic moment may be considered to arise only from unpaired spins. The spin-only magnetic moment t may be written:
The number of unpaired spins n by the equation:
The spin-only results are shown in Table. The simple spin-only moments shown in table give good agreement with many high-spin complexes of first row elements
Spin only magnetic moments for numbers of unpaired electrons:
|
Number of unpaired electrons n |
Magnetic moment μS (BM) |
Total spin quantum number S |
|
1 |
1.73 |
1/2 |
|
2 |
2.83 |
2/2 = 1 |
|
3. |
3.87 |
3/2 |
|
4. |
4.90 |
4/2 = 2 |
|
5. |
5.92 |
5/2 |
Magnetic moments of some first row complexes:
|
Ion |
Number of unpaired electrons |
Experimental magnetic moment (BM) |
Calculated magnetic moment spin only formula μs (BM) |
|
Ti`3+ |
1 |
1.7-1.8 |
1.73 |
|
V3+ |
2 |
2.8-3.1 |
2.83 |
|
Cr3+ |
3 |
3.7-3.9 |
3.87 |
|
Cr2+, Mn3+ |
4 |
4.8-4.9 |
4.90 |
|
Mn2+, Fe3+ |
5 |
5.7–6.0 |
5.92 |
|
Fe2+ |
4 |
5.0-5.6 |
4.90 |
|
Co2+ |
3 |
4.3-5.2 |
3.87 |
|
Ni2+ |
2 |
2.9–3.9 |
2.83 |
|
Cu2+ |
1 |
1.9-2.1 |
1.73 |
Illustration 9: Of the ions Co2+, Sc3+ and Cr3+, which ones will give coloured aqueous solutions and how will each of them respond to a magnetic field and why?
(Atomic numbers: Co = 27, Cr = 24).
Solution: Co2+ and Cr3+ have unpaired electrons in the 3d subshell. Hence, they will give coloured solutions and attracted by the magnetic field. Sc3+ has no unpaired electron. Hence, it will be repelled by the external magnetic field.
Catalytic Properties:
Transition metals and their compounds have catalytic properties.
|
Pt |
Formerly used for SO2 →SO3 in the contact process for making H2SO4. |
|
Pt |
Is increasingly being used in three stage-convertors for cleaning car exhaust fumes. |
|
Pt/Rh |
Formerly used in the Ostwald process for making HNO3 to oxidize NH3 to NO. |
|
Cu |
Is used in the direct process for manufacture of (CH3)2SiCl2 used to make silicones. |
|
Cu/V |
Oxidation of cyclohexanol/cyclohexanone mixtures to adipic acid which is used to make nylon-66. |
|
CuCl2 |
Deacon process of making Cl2 from HCl. |
|
Ni |
Raney nickel, numerous reduction processes (e.g., manufacture of hexamethylenediamine, production of H2 from NH3, reducing anthraquinone to anthraquinol in the production of H2O2) |
|
Ni complexes |
Reppe synthesis (polymerization of alkynes, e.g., to give benzene or cyclooctatetraene. |
|
TiCl3 |
Used as the Ziegler-Natta catalyst in the production of polythene |
|
V2O5 |
Converts SO2 to SO3 in the Contact process for making H2SO4 |
|
MnO2 |
Used as a catalyst to decompose KClO3 to give O2. |
|
Fe |
Promoted iron is used in the Haber—Bosch process for making NH |
|
FeCl3 |
Used in the production of CCl4 from CS2 and Cl2 |
|
FeSO4 & H2O2 |
(Fenton’s reagent) For oxidizing alcohol to aldehyde |
|
PdCl2 |
Wacker process for converting C2H4 + H2O + PdCl2 → CH3CHO + 2HCl + Pd. |
|
Pd |
Used for hydrogenation (e.g. phenol to cylohexanone) |
|
Pt/PtO |
Adams catalyst, used for reductions |
In some cases the transition metals with their variable valency may form unstable intermediate compounds. In other cases the transition metal provides a suitable reaction surface. This is how they behave as catalysts.
Enzymes are catalysts that enhance the rates of specific reactions. They are proteins and are produced by living cells from amino acids. They work under mild conditions and often give 100% yields and may speed a reaction by 106 or 1012 times.
Some enzymes require the presence of metal ions as cofactors. and these are called metalloenzymes. Almost of metalloenzymes contain a transition metal. Some metalloenzymes are listed in following table:
|
Metal |
Enzyme/metalloprotein |
Biological function |
|
Mo |
Xanthine oxidase Nitrate redcatse |
Metabolism of purines Utilization of nitrates |
|
Mn(II) |
Arginase Phosphotransferases |
Urea formation Adding or removing |
|
Fe(II) or (III) |
Aldehyde oxidase Catalase Feroxidase Cytochromes Ferredoxin (Haemoglobin) Succinic dehydrogenase |
Oxidation of aldehydes Decomposes H2O2 Decomposes H2O2 Electron transfer Photosynthesis O2 transport in higher animals Aerobic oxidation of carbohydrates |
|
Fe and Mo |
Nitrogenase |
Fixation of dinitrogen |
|
Co |
Glutamic mutase Ribonucleotide reductase |
Metabolism of amino acids Biosynthesis of DNA |
|
Cu(I) or (II) |
Amine oxidases Ascorbate oxidase Cytochrome oxidase Galactose oxidase Lysine oxidase Dopamine hydroxylase
Tyrosinase Ceruloplasmin (Haemocyani) Plastocyanin |
Oxiation of amines Oxidation of ascorbic acid Principal terminal oxidase Oxidation of galactose Elasticity of aortic walls Producing noradrenaline to generate nerve impulses in the brain Skin pigmentation Utilization of Fe O2 transport in invertebrates Photosyntehsis |
|
Zn(II) |
Alcohol dehydrogenase Alkaline phosphatase Carbonic anhydrase Carboxypeptidase |
Metabolism of alcohol Releasing Regulation of pH and CO2 formation Digestion of proteins |
Nonstoichiometry:
A further feature of the transition elements is that they sometimes form nonstoichiometric compounds. These are compounds of indefinite structure and proportions. For example, iron(II) oxide FeO should be written with a bar over the formula to indicate that the ratio of Fe and O atoms is not exactly 1: 1. Analysis shows that the formula varies between Fe0.94O and Fe0.84O. Vanadium and selenium form a series of compounds ranging from VSe0.98 to VSe2. These have been given the formulae:
Nonstoichiometry is shown among transition metal compounds of the Group 16 elements (O, S, Se, Te). It is mostly due to the variable valency of transition elements. For example copper is precipitated from a solution containing Cu2+ by passing in H2S. The sulphide is completely insoluble, but this is not used as a gravimetric method for analysing for Cu2S because the precipitate is a mixture of CuS and Cu2S. Sometimes nonstoichiometry is caused by defects in the solid structures.
Extraction of iron:
Occurrence: Iron is the fourth most abundant element and the second most abundant metal in nature. It occurs in combined state.
Types of ores:
a. Oxide ores:
(i) Magnetite (Fe3O4) It is usually black in colour. It is richest ore of iron and contains upto 70% of the metal.
(ii) Haematite (Fe2O3) Fe It is usually red in colour.
(iii) Limonite or hydrated ferric oxide (Fe2O3.3H2O) It has yellow, brown or red colour.
b. Carbonate ore:
Siderite or Spathic iron (FeCO3) It is also called clay- iron stone due to the presence of excess of clay in it.
c. Sulphide ores:
(i) Iron pyrites (FeS2) it is mainly used for the manufacture of sulphur dioxide (sulphuric acid). It is not used for extraction of iron.
(ii) Chalcopyrites (CuFeS2) It is used for the extraction of copper.
Iron ores have been reported at many places especially in England, Sweden, Germany, U.S.A., Russia, Belgium, France, Canada and India. Huge deposits of red haematite are available in India in Mayurbhanj (Orissa), Singhbhum (Bihar), Madhya Pradesh and Mysore (Karnataka). On the whole, India possesses about one fourth of the total world reserves of iron ore. Iron is extracted from its oxide ores especially from the magnetite, haematite and limonite. The extraction involves the following steps:
(1) Concentration of ore: The concentration of ore is done by gravity process. The ore is crushed to small pieces and washed with water to remove silicious impurities. The washed ore is then subjected to electromagnetic separation.
(2) Calcination and roasting: The concentrated ore is heated in excess of air. This treatment produces the following results.
(a) Moisture and carbon dioxide are removed.
2Fe2O3.3H2O → 2Fe2O3 + 3H2O
FeCO3 → FeO + CO2
(b) Sulphur, arsenic, etc., are oxidised to their oxides and are, thus, removed as volatile gases.
S + O2 → SO2
4As + 3O2 → 2As2O3
(c) Ferrous oxide is oxidised to ferric oxide.
(d) The entire mass becomes porous.
(Smelting: The calcined ore is mixed with limestone (CaCO and coke in the ratio of S : I : 4 and introduced in a blast furnace for smelting.
Blast furnace: Blast furnace is a chimney like tall steel structure lined with fire bricks, 25 – 60 metre high and 8-10 metre in diameter at its widest part (see the following figure). The furnace has three main parts:
(i) A double cup and cone arrangement: At the top, the furnace has a hopper which rests over a cup and cone arrangement. By this arrangement charge is introduced into the furnace ant also the arrangement prevents the exit of gases during charging.
(ii) Shaft: It is formed by joining two cones, the upper one is called the body and the lower one is called bosh. At the
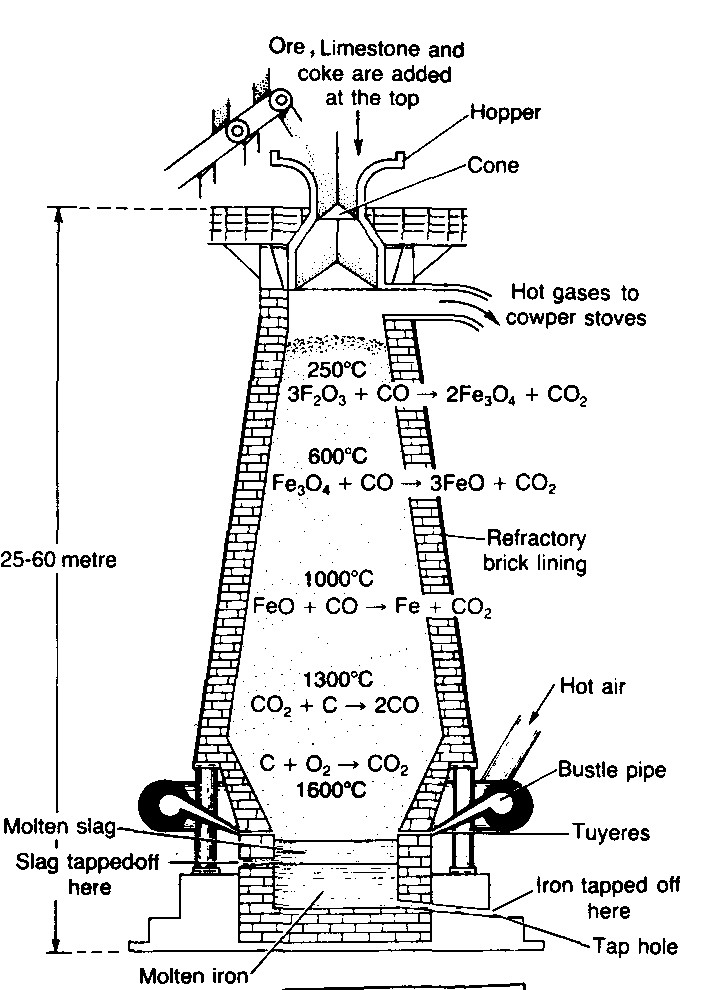
upper part, there is a hole, through which the hot burnt mixture of waste gases escape. In the lower part, there are openings for the insertion of water cooled pipes called tuyeres through which a blast of hot air is sent upward in the furnace.
(iii) Hearth: This is the lowest part of the furnace and serves as a large crucible. It is provided with two holes one for the removal of slag and other for molten metal.
The charge is introduced in the furnace by lowering the cup and cone arrangement and at the same time the furnace is lit and a blast of hot air is sent upwards through the tuyeres. The temperature varies from 1600°C to 250°C in the furnace from bottom to top. On the basis of variation in temperature, there are four zones where different chemical changes occur. The four zones are
a. Combustion zone: This is the lowest part of the furnace above hearth where the temperature is about 15000 – 16000 In this zone carbon burns in presence of hot air producing carbon dioxide and a lot of heat.
C + O2 → CO2 + 97.0 k.cals.
Carbon dioxide rises upwards and meets with red hot coke. It is reduced to carbon monoxide.
(iii) Limonite or hydrated ferric oxide, Fe2O3.3H2O. It has yellow, brown or red colour.
b. Carbonate ore: Siderite or Sepathic iron, FeCO3 It is also called clay- iron stone due to the presence of excess of clay in it.
c. Sulphide ores:
(i) Iron pyrites, FeS2 It is mainly used for the manufacture of sulphur dioxide (sulphuric acid). It is not used for extraction of iron.
(ii) Chalcopyrites, CuFeS2 It is used for the extraction of copper.
Iron ores have been reported at many places especially in England, Sweden, Germany, U.S.A.. Russia, Belgium, France, Canada and India. Huge deposits of red haematite are available in India in Mayurbhanj (Orissa), Singhbhum (Bihar), Madhya Pradesh and Mysore (Karnataka). On the whole, India possesses about one fourth of the total world reserves of iron ore.
Iron is extracted from its oxide ores especially from the magnetite, haematite and limonite ores. The extraction involves the following steps.
(I) Concentration of ore: The concentration of ore is done by gravity process. The ore is crushed to small pieces and washed with water to remove silicious impurities. The washed ore is then subjected to electromagnetic separation.
(ii) Calcination and roasting: The concentrated ore is heated in excess of air. This treatment produces the following results.
(a) Moisture and carbon dioxide are removed.
(b) Sulphur, arsenic, etc., are oxidised to their oxides and are, thus, removed as volatile gases.
S + O2 → SO2
4As + 3O2 → 2As2O3
(c) Ferrous oxide is oxidised to ferric oxide.
(d) The entire mass becomes porous.
(3) Smelting: The calcined ore is mixed with limestone (CaCO3) and coke in the ratio of 8 : I : 4 and introduced in a blest furnace for smelting.
Blast furnace: Blast furnace is a chimney like tall steel structure lined with fire bricks, 25-60 metre high and 8-10 metre in diameter at its widest part (Fig. 14.1). The furnace has three main parts:
(1) A double cup. and cone arrangements: At the top, the furnace has a hopper which rests over a cup and cone arrangement. By this arrangement charge is introduced into the furnace an also the arrangement prevents the exit of gases during charging.
(2) Shaft: It is formed by joining two cones, the upper one is called the body and the lower one is called bosh. At the upper part, there is a hole, through which the hot burnt mixture of waste gases escape. In the lower part, there are openings for the insertion of water cooled pipes called tuyeres through which a blast of hot air is sent upward in the furnace.
(3) Hearth : This is the lowest part of the furnace and serves as a large crucible. It is provided with two holes one for the removal of slag and other for molten metal.
The charge is introduced in the furnace by lowering the cup and cone arrangement and at the same time the furnace is lit and a blast of hot air is sent upwards through the tuyeres. The temperature varies from 1600°C to 250°C in the furnace from bottom to top. On the basis of variation in temperature, there are four zones where different chemical changes occur. The four zones are
1. Combustion zone: This is the lowest part of the furnace above hearth where the temperature is about 1500°—I 600°C. In this zone carbon burns in presence of hot air producing carbon dioxide and a lot of heat.
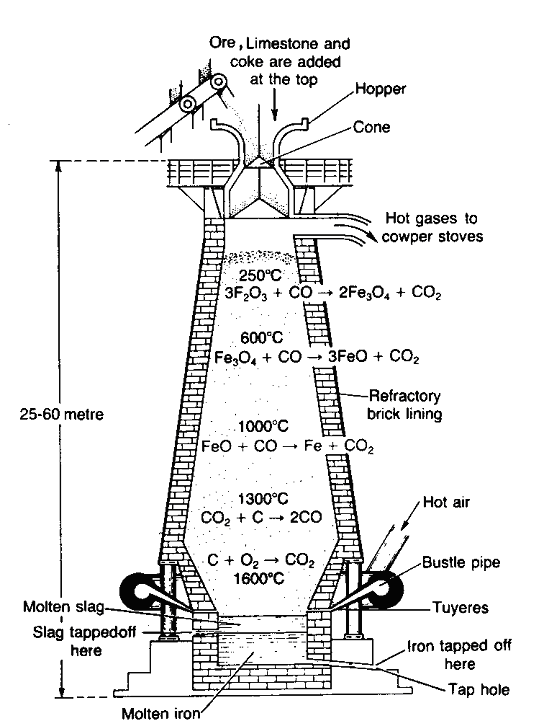
C + O2 → CO2 + 97.0 kcal
Carbon dioxide rises upwards and meets with red hot coke. It is reduced to carbon monoxide.
2. Reduction zone: This is the uppermost part of the furnace. The temperature varies from 250°C to 700°C. The oxide ore is reduced to iron in this zone. The reduction takes place through the following stages:
Iron formed is called spongy iron. In the reduction reactions heat is also evolved which decomposes part of carbon monoxide into carbon.
3. Slag formation zone: This is the central zone where the temperature varies from 800-1000°C. The limestone present in the charge decomposes into calcium oxide and carbon dioxide.
The calcium oxide acts as a flux as it combines with silica present as an impurity (gangue) to form fusible slag of CaSiO3.
CaO + SiO2 → CaSiO3
Silicates, phosphates and manganates present as impurities in ore, are reduced to Si, P and Mn, respectively.
P4O10 + 10C → 4P + 10CO
SiO2 + 2C → Si + 2CO
MnO2 + 2C → Mn + 2CO
These are partly absorbed by iron (spongy) and partly by slag.
2Ca3 (PO3)2 + 3SiO2 + 10C → 3 (2CaO-SiO2) + 4P + 10CO
3Fe + P → Fe3P
4. Zone of fusion: This is a zone just above the zone of combustion. The temperature ranges between 1200—I 500°C. The spongy iron which has absorbed already C, Si, P, Mn, etc., melts at 1300°C and collects at the bottom of the hearth. The slag which being lighter floats over the molten iron and prevents the oxidation of molten metal. The slag and molten metal are removed from their respective holes. The molten metal is run into moulds and is allowed to solidify.
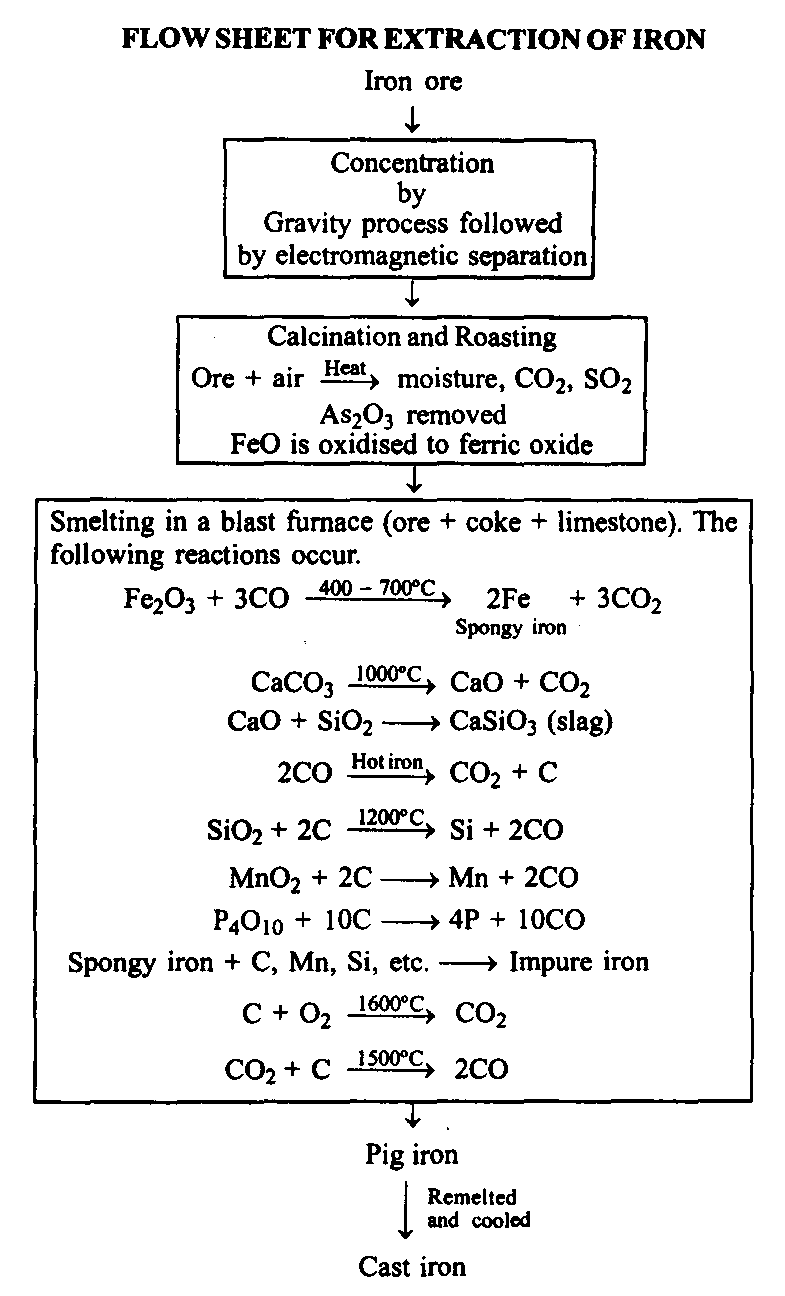
(Fe = 93%; C = 5% and impurities of Mn, 1’, Si, etc. = 2%) Iron obtained from the blast furnace is called pig Iron. It contains 93% iron, 5% carbon and rest silicon, manganese, phosphorus, etc., as impurities. The pig iron is remelted and cast or poured into moulds. This is known as cast iron. Hence, after remelting the pig iron becomes cast iron.
When the molten pig iron is cooled at once, the iron is called white cast Iron, which contains carbon in the form of cementite, Fe and when the molten pig iron is cooled slowly and slowly, the iron is called as grey cast iron, which contains carbon in the form of graphite.
Types of iron:
There are three commercial varieties of iron depending on their carbon content.
( Cast iron: It is the most impure form of iron and contains the highest percentage of carbon from 2.5 to 5 per cent and about 2 per cent of other impurities like Si, P, Mn and S. Cast iron is of two types:
(a) White cast iron: Carbon is present in the form of cementite, Fe
(b) Grey cast iron: Carbon is present in the form of graphite. Cast iron melts at about 1250°C (due to presence of impurities) whereas pure iron melts at 1530°C. The molten cast iron expands on solidification and hence it produces good castings. Various articles such as stoves, pipes, radiators, railway sleepers, gutter pipes, toys, etc., are prepared from cast iron. Cast iron does not rust easily and neither be tempered. Due to high carbon content, it is hard and brittle and cannot be welded. It has very little ductility and thus cast iron is not suitable for forging.
(if) Wrought iron: It is the purest form of iron. It contains the lowest percentage of carbon from 0.1 to 0.25 per cent and 0.3 per cent other impurities. It is manufactured from cast iron by puddling process.
Wrought iron is manufactured in a special type of reverberatory f called puddling furnace, the hearth of which is lined with haematite, Fe The cast iron is melted on the hearth of the furnace by the hot gases and stirred with long iron rods. The impurities of cast iron are rapidly oxidised by oxygen of haematite (lining). Oxides of carbon and sulphur being volatile escape while those of Mn, P and Si form slag.
3C + Fe2O3 → 2Fe + 3CO
3Si + 2Fe2O3 → 3SiO2 + 4Fe
3Mn + fE2O3 → 3MnO + 2Fe
MnO + SiO2 → MnSiO3 (slag)
6P + 5Fe2O3 → 3P2O5 + 10Fe
P2O3 + Fe2O3 → 2FePO4 (slag)
With the removal of impurities, the melting point of the metal rises and it becomes a semi-solid mass. The semi-solid mass is taken out in the form of balls and is beaten under steam hammers to squeeze out as much of slag as possible. This produces almost pure iron known as wrought iron.
Properties: (i) Wrought iron is extremely tough, highly malleable and ductile. (ii) It softens at about 1000°C and then it can be forged and welded. (iii) On account of the presence of very small percentage of slag, it has fibrous structure and thus, can withstand high stresses. (iv) Wrought iron is resistant towards rusting and corrosion.
Uses: It is used to make chains, nails, hooks, bolts, agricultural implements, electromagnets, bars, wires, etc.
(iii) Steel: This is the most important commercial variety of iron. The percentage of carbon in this form of iron is midway between that of cast iron and wrought iron, i.e., 0.25 to 2 per cent. There are many varieties of steel depending on the amount of carbon present in it.
(a) Mild steels: These contain low percentage of carbon. Such steels show the properties of wrought iron along with elasticity and hardness.
(b) Hard steels: These contain high percentage of carbon. They are hard and brittle.
(c) Special steels or alloy steels: Steel mixed with small amount of nickel, cobalt, chromium, tungsten, molybdenum. manganese, etc., acquires special properties. Such products are called special steels or alloy steels. Some important alloy steels are listed below:
|
Name of the alloy steel |
Metal added |
Properties |
Uses |
|
1. Invar |
36% Ni |
Coefficient of expansion is very small |
Measuring tapes pendulums |
|
2. Chromevandium steel |
1% Cr; 0.15%V |
High tensile strength |
Springs, shafts, axles |
|
3. Manganese steel |
12-15% Mn |
Hard and tough |
Rock crushing machinery, almirah, helmets |
|
4. Stainless steel |
11.5% Cr; 2% Ni |
Resists corrosion |
Common articles |
|
5. Tungsten steel |
14-20% W; 3-8 Cr |
Very hard |
High speed tools |
Manufacture of steel: Many methods are used for the manufacture of steel. Some are described below:
The Bessemer’s process: The process is based on the fact that impurities of pig iron are completely oxidised in
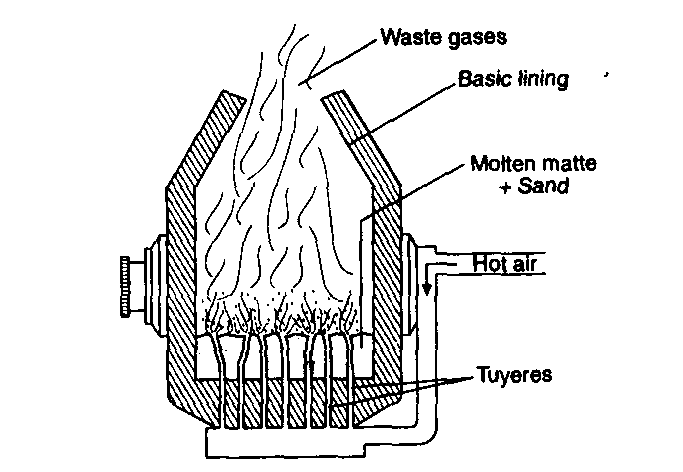
presence of hot air blast, i.e., virtually wrought iron is obtained. This is then mixed with a known amount of spiegeleisen, an alloy of iron, manganese and carbon to obtain steel.
The process is carried out in Bessemer converter lined with silica bricks. The molten pig iron is introduced in the converter and a blast of hot air is blown through it from the bottom and keeping the mouth of the converter vertically upwards. Silica and manganese present in pig iron are first oxidised and then combine to form slag.
Si + O2 → SiO2
2Mn + O2 → 2MnO
MnO + SiO2 → MnSiO3
In the end, carbon is oxidised to carbon monoxide which bums with blue flame at the mouth of the converter. Some iron is also oxidised which converts the carbon into carbon monoxide.
4Fe + 3O2 → 2Fe2O3
Fe2O3 + 3C → 2Fe + 3CO
When whole of the carbon is oxidised, the blue flame suddenly dies out. The air supply is stopped for a while and the requisite amount of spiegeleisen is added. The blast is continued just for a moment to ensure complete mixing. The resulting product is the manganese steel.
When cast iron or pig iron contains phosphorus as an impurity, a basic lining of CaO or MgO is used in the Bessemer converter. Phosphorus is oxidised to P4O10 which combines with CaO to form calcium phosphate as slag.
4P + 5O2 → P4O10
6CaO + P4O10 → 2Ca3(PO4)2
This slag is used as a fertilizer and known as Thomas slag.
(ii) Open hearth process or Siemens-Martin process:
This is the modern process and the furnace used consists of an open hearth. The hearth is lined with silica or calcined dolomite (CaOMgO) depending upon the nature of impurities present in pig or cast iron. Silica lining is used if the impurities are manganese, silicon, etc., and calcined dolomite lining is used if much of phosphorus is present. A high temperature of about 1500°C is generated by burning producer gas which works on regenerative system of heat economy (Fig).
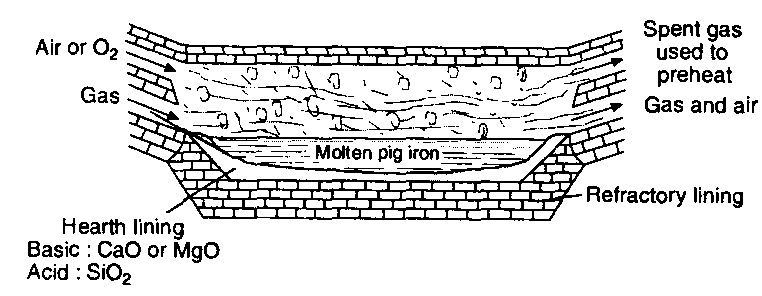
The charge consists of pig or cast iron, iron scrap, iron ore (haematite) and lime. The charge is heated on the hearth of the furnace. The impurities are oxidised by iron ore.
3Si + 2Fe2O3 → 4Fe + 3SiO2
3Mn + Fe2O3 → 2Fe + 3MnO
MnO + SiO2 → MnSiO3 (slag)
3C + Fe2O3 → 2Fe + 3CO
12P + 10Fe2O3 → 3P4O10 + 20Fe
Samples of steel are drawn from time to time and tested for carbon content. Finally spiegeleisen (an alloy of iron, manganese and carbon) is added to the molten mass to obtain desired steel. The process takes about 8 to 10 hours for completion. The process takes longer time than Bessemer’s process but it has following advantage over the Bessemer’s process.
(a) The temperature can be controlled as the heating is done externally.
(b) As it is a slower process, it can be controlled in better way. The composition and quality can be well controlled.
(c) The loss of iron in this process is only 4% while the loss is about 15% in Bessemer’s process.
(d) In this process scrap iron is re-used.
(e) This yields better quality of steel.
(f) A considerable economy of the fuel is achieved by using the regenerative system.
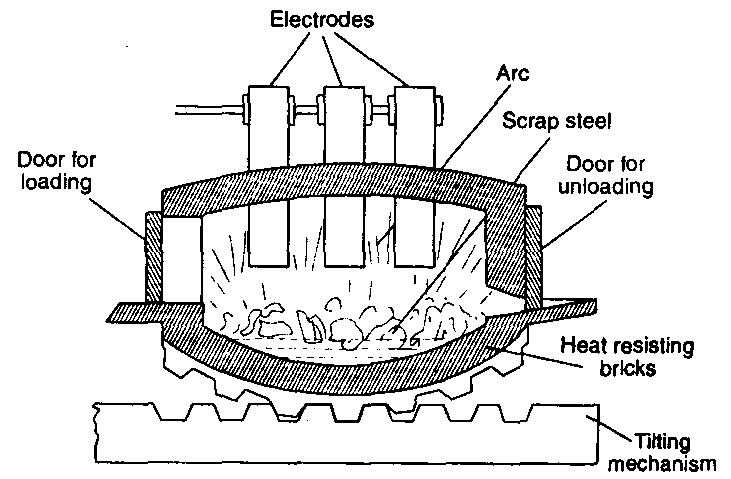
(ill) The electric process : This process is similar to open hearth process with a difference that heating is done electrically. The steel of much better quality can be obtained but the process is rather costly.
The process consists in heating of the charge having pig or cast iron, scrap iron, iron ore (haematite) and lime in an electric furnace using vertical carbon electrodes. When the arc is struck between the electrodes, the high temperature of about 2000°C generated, melts the charge and chemical reactions start instantaneously. The impurities are oxidised by iron ore in the same fashion as in the open hearth process.
This method is especially useful for the production of the alloy steels.
Heat treatment of steels: The properties of steel depend on three factors
(i) Carbon content: With the increase of carbon content the hardness and tensile strength of the steel increases while ductility decreases.
Comparison of Cast Iron, Wrought Iron and Steel:
|
Property |
Cast iron |
Wrought iron |
Steel |
|
1. Chemical composition |
Iron 93 – 95% |
Iron 99.5 – 99.8% |
Iron 99.5-98% |
|
2. Melting point |
Lowest about 12000C |
Highest about 15000C |
Between 1300-14000C |
|
3. Hardness |
Very hard |
Soft |
Medium hardness |
|
4. Malleability |
Brittle |
Malleable |
Malleable and brittle |
|
5. Welding |
Brittle |
Malleable |
Malleable and brittle |
|
6. Tempering |
Cannot be tempered |
Cannot be tempered |
Can be tempered |
|
7. Magnetisation |
Cannot be permanently magnetized |
Magnetisation is not permanent but easy |
Can be permanently magnetized |
|
8. Structure |
Crystalline |
Fibrous |
Granular |
(ii) Presence of other metals: The presence of Si gives steel a fibrous structure. Presence of Mn produces elasticity and increases tensile strength. Cr imparts resistance to chemical action.
(iii) Heat treatment: The hardness and elasticity of the steel can be changed by heating the steel at different temperatures and then allowing to cool it in different ways. The following heat treatments are given to steel
(a) Annealing: It is a process of heating steel to bright redness and then cooling it very slowly. This treatment makes the steel soft and ductile. This type of steel is used in fabrication process.
(b) Quenching: it is a process of heating steel to bright redness and then cooling it suddenly by plunging it in water or oil. Such a steel is extremely hard and brittle. It has very low elasticity.
(c) Tempering: It is a process of heating the quenched steel to a temperature much below redness and then cooling it slowly. Such steel is neither so hard nor so brittle. In the process of tempering, a thin film of the oxide is formed on the surface of steel. The colour of the oxide film depends on the temperature at which the quenched steel is heated.
Temperature range Colour of the oxide film
200 Yellow
225-270°C Brown
300°C Blue
The surface treatment of the steel is done by the following two processes:
(a) Case hardening: The process of producing a thin coating of hardened steel on the surface of the mild steel is called case hardening. This is done by heating the mild steel with charcoal and then plunging into oil. This produces a thin coating of hardened steel on the surface. Such a steel becomes resistant to wear and tear.
(b) Nitriding: The process of producing a hard coating of iron nitride on the surface of steel is called nitriding. Steel is heated in the atmosphere of dry ammonia at 500—600°C for about 3 to 4 days when a hard coating of iron nitride is produced on the surface.
Passivity of Iron:
The following are the common properties of iron.
(a) It evolves hydrogen gas, when made to react with dilute HCI or dilute H2SO4.
(b) It precipitates silver from silver nitrate solution and copper from copper sulphate solution.
But if a piece of iron is first dipped in concentrated nitric acid for sometime and then made to react with the above reagents, neither hydrogen is evolved nor silver or copper are precipitated. Thus, iron by treatment with concentrated nitric acid has lost its usual properties or it has been rendered inert or passive. Such behaviour is not only shown by iron but also by many other metals like Cr, Co, Ni, Al, etc. This phenomenon is known as passivity and the chemical substances which bring passivity are called passivators.
“The inertness exhibited by metals under conditions when chemical activity is to be expected is called chemical passivity.” Iron can be rendered passive by other oxidising agents like chromic acid, KMnO4 conc. H etc. The passivity of the iron is believed to be due to formation of an extremely thin film (invisible) of oxide on the surface of iron. Passive iron can be made active by scratching or heating in a reducing atmosphere of H2 or CO2 or heating in HNO3 upto 75°C.
Compounds of Iron:
Ferrous sulphate (Green vitriol), FeSO4)
This is the best known ferrous salt. It occurs in nature as copperas and is formed by the oxidation of pyrites under the action of water and atmospheric air.
2FeS2 + 7O2 + 2H2O → 2FeSO4 + 2H2SO4
It is commonly known as harakasis.
Preparation: (i) it is obtained by dissolving scrap iron in dilute sulphuric acid.
Fe + H2SO4 → FeSO4 + H2
The solution is crystallised by the addition of alcohol as ferrous sulphate is sparingly soluble in it.
(ii) It can also be prepared in the laboratory from the Kipp’s waste. The excess of sulphuric acid is neutralised by heating with a small quantity of iron fillings. The solution is then crystallised.
Manufacture: Commercially, ferrous sulphate is obtained by the slow oxidation of iron pyrites in the presence of air and moisture. The pyrites are exposed to air in big heaps.
2FeS2 + 2H2O + 7O2 → 2FeSO4 + 2H2SO4
The free sulphuric acid is removed by the addition of scrap iron. On crystallisation green crystals are obtained.
Properties: (i) Hydrated ferrous sulphate (FeSO27H2O) is a green crystalline compound. Due to atmospheric oxidation, the crystals acquire brownish-yellow colour due to formation of basic ferric sulphate.
(ii) Action of heat: At 300°C, it becomes anhydrous.
The anhydrous ferrous sulphate is colourless. The anhydrous salt when strongly heated, breaks up to form ferric oxide with the evolution of SO2 and SO3.
(ii) Action of heat: At 300°C, it becomes anhydrous.
The anhydrous ferrous sulphate is colourless. The anhydrous salt when strongly heated, breaks up to form ferric oxide with the evolution of SO2 and SO3.
(iii) The aqueous solution of ferrous sulphate is slightly acidic due to its hydrolysis.
(iv) Ferrous sulphate is a strong reducing agent.
(a) It decolourises acidified potassium permanganate.
(b) It turns potassium dichromate (acidi&d) green as dichrom is reduced to chroinic salt (green).
(c) it reduces gold chloride to gold.
AuCl3 + 3FeSO4 → Au + Fe2(SO4)3 + FeCl3
(d) it reduces mercuric chloride to mercurous chloride.
(v) A cold solution of ferrous sulphate absorbs nitric oxide forming dark brown addition compound, nitroso ferrous sulphate.
The NO gas is evolved when the solution is heated.
(vi) It forms double sulphate of the composition R2SO4.FeSO4.6H2O where R = an alkali metal or radical.
(NH4)2SO4.FeSO4.6H2O (ferrous ammonium sulphate) is known as Mohr’s salt.
(vii) It combines with potassium cyanide (excess) forming potassium ferrocyanide, K4Fe(CN)6.
Uses : (i) Ferrous sulphate is used for making blue black ink. The ink is prepared by mixing a solution of tannin and ferrous sulphate. A colourless iron salt of tannic acid is formed. A blue dye (usually indigo) is added. The writing with this ink is blue but soon on exposure to air, it becomes black on account of oxidation of colourless ferrous salt into black ferric salt.
(ii) It is used as a mordant in dyeing.
(iii) It is also used as an insecticide in agriculture.
(iv) It is employed as a laboratory reagent and in the preparation of Mohr’s salt.
Ferrous ammonium sulphate (Mohr’s salt)
(NH4)2 SO4. FeSO4. 6H2O:
Preparation: The double salt is best prepared by making saturated solutions of pure ferrous sulphate and pure ammonium sulphate in air free distilled water at 40°C. Both the solutions are mixed and allowed to cool. Generally, few drops of sulphuric acid and a little iron wire are added before crystallisation as to prevent oxidation of ferrous sulphate into ferric sulphate. The salt is obtained as pale green crystals.
Properties: It is pale green crystalline compound which does not effloresce like ferrous sulphate. It is less readily oxidised in the solid state. It is, therefore, a better volumetric reagent in preference to ferrous sulphate. Chemically, it is similar to ferrous sulphate. All the chemical reactions observed in the case of ferrous sulphate are given by ferrous ammonium sulphate.
Ferric chloride, FeCI3
This is the most important ferric salt. It is known in anhydrous and hydrated forms. The hydrated form consists of six water molecules, FeCl3CH2O.
Preparation: (i) Anhydrous ferric chloride is obtained by passing dry chlorine gas over heated iron fillings
The vapours are condensed in a bottle attached to the outlet of the tube.
2Fe + 3Cl2 → 2FeCl2
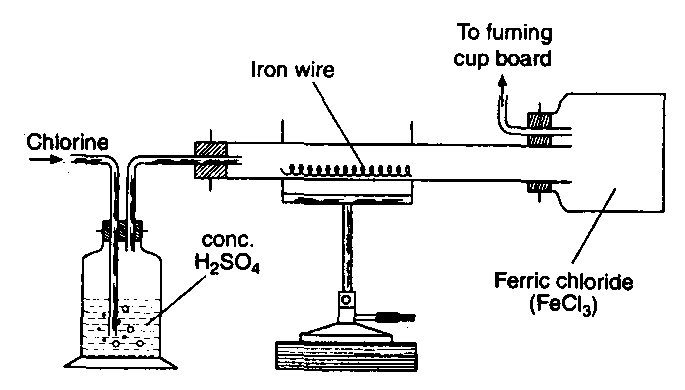
(ii) Hydrated ferric chloride is obtained by the action of hydrochloric acid on ferric carbonate, ferric hydroxide or ferric oxide.
The solution on evaporation and cooling deposits yellow crystals of hydrated ferric chloride, FeCl3
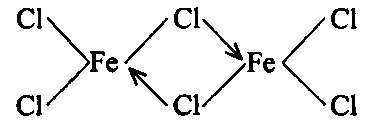
Properties: (i) Anhydrous ferric chloride is a dark red deliquescent solid. It is sublimed at about 300°C and its vapour density corresponds to dimeric formula, Fe The dimer dissociates at high temperatures to FeCl3 The dissociation into Fed is complete at 750°C. Above this temperature it breaks into ferrous chloride and chlorine.
(ii) Anhydrous ferric chloride behaves as a covalent compound as it is soluble in non-polar solvents like ether, alcohol, etc. It is represented by chlorine bridge structure.
(iii) It dissolves in water. The solution is acidic in nature due to its hydrolysis as shown below
The solution is stabilised by the addition of hydrochloric acid to prevent hydrolysis.
(iv) Anhydrous ferric chloride absorbs ammonia.
(v) Ferric chloride acts as an oxidising agent.
(a) It oxidises stannous chloride to stannic chloride.
(b) It oxidises SO2 to H2SO4.
(c) It oxidises H2S to S.
(d) It liberates iodine from KI
(e) Nascent hydrogen reduces FeCl3 into FeCI2.
(vi) When ammonium hydroxide is added to the solution of ferric chloride, a reddish-brown precipitate of ferric hydroxide is formed.
(vii) When a solution of thiocyanate ions is added to ferric chloride solution, a deep red colouration is produced due to formation of a complex salt.
or
(viii) Ferric chloride forms a complex, pussian blue with potassium ferrocyanide.
(ix) On heating hydrated ferric chloride FeCl3.6H2O, anhydrous ferric chloride is not obtained. It is changed to Fe2O3 with evolution of H2O and HCI.
Hydrated ferric chloride may be dehydrated by heating with thionylchloride.
Uses:
(i) The alcoholic solution is used in medicine under the name tincture fern perchloride.
(ii) It is used as a laboratory reagent in the detection of acetates and phenols and also as an oxidising agent.
(iii) It is used for making prussian blue.
Corrosion of iron:
Corrosion Is defined as the gradual transformation of a metal Into its combined state because of the reaction with the environment. Metals are usually extracted from their ores. Nature tries to convert them again into the ore form. The process by which the metals have the tendency to go back to their combined state, is termed corrosion.
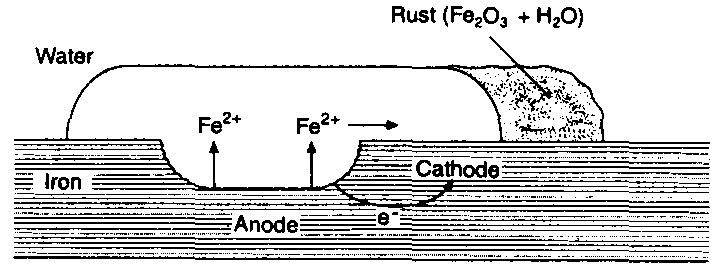
When iron is exposed to moist air, it is found covered with a reddish-brown coating which can easily be detached. The reddish-brown coating is called ‘rust’. Thus, the corrosion of iron or formation of the rust is called rusting. The composition of the rust is not certain but it mainly contains hydrated ferric oxide, 2Fe2O3.3H2O together with a small quantity of ferrous carbonate. The rust is formed by the action of water on iron in presence of dissolved oxygen and carbon dioxide. It has been observed that impure iron is more prone to rusting.
The following are the favourable conditions for the rusting of iron:
(i) Presence of moisture
(ii) Presence of a weakly acidic atmosphere
(iii) Presence of impurity in the iron.
Various theories have been proposed to explain the phenomenon of rusting of iron but the accepted theory is the modern electrochemical theory. When impure iron comes in contact with water containing dissolved carbon dioxide, a voltaic cell is set up. The iron and other impurities act as electrodes while water having dissolved oxygen and carbon dioxide acts as an electrolyte. Iron atoms pass into solution as ferrous ions.
Fe → Fe2+ + 2e
Iron, thus, acts as anode.
The impurities act as cathode. At the cathode, the electrons are used in forming hydroxyl ions.
H2O + O + 2e → 2OH–
In presence of dissolved oxygen, ferrous ions are oxidised to ferric ions which combine with hydroxyl ions to form ferric hydroxide.
Fe3+ + 3OH– → Fe(OH)3
[2Fe2+ + H2O + O → 2Fe3+ + 2OH–]
Corrosion or rusting is a surface phenomenon and thus, the protection of the surface prevents the corrosion. Iron can be protected from the rusting by use of following methods
(i) Applying paints, lacquers and enamels on the surface of iron.
(ii) By forming a firm and coherent protective coating of ferrosoferric oxide. This is done by passing steam over hot iron.
(iii) By coating a thin film of zinc, tin, nickel, chromium, aluminium, etc.
Oxides & Oxometal ion:
Oxides are generally formed by the reaction of oxygen with metal at high temperature. All metals (except Se) form ionic oxide with general formula MO. Transition metals show their highest oxidation state in their oxides which are equal to their group number but up to group 7, (eg., Mn2O) thereafter, no higher oxides are known (e.g., for Fe, Fe2O3). Besides oxides, oxocations also exist e.g., VIII in VO2+, VII in VO2+ and TiII in TiO2+.
With increasing Ox. no., there is an increase in the covalent nature i.e., the ionic character decreases. For example Mn2O7 is a covalent liquid higher oxides have predominant acidic character – as when dissolved in aq. medium they give acids: Mn2O7 gives HMnO4 (permanganic acid) and CrO3 gives H2CrO4 (chronic acid) and H2Cr2O7 (dichromic acid) V2O5 is however amphoteric, although in majority of reactions it is acidic and gives VO43– as well as VO2+ salts behaving as a base. In fact there is a gradual change from basic to less basic in vanadium from V2O3 to V2O4. The latter dissolves in acids to give VO2+ salts. Lower oxides of Cr and Mn (e.q., CrO and MnO, Mn2O3) are basic those with intermediate OX states (between highest & lowest) are amphoteric (e.g., Cr2O3 and MnO2)
Potassium Dichromate, K2Cr2O7:
Preparation: It is the most important compound of Cr (VI). It is manufactured from chromite ore. Chromite ore is first converted into sodium dichromate.
It is manufactured from chromite ore [FeCr2O4] which is first converted into sodium dichromate as follows:
Sodium dichromate is hygroscopic and more soluble than poto dichromate
The hot saturated solution of Na2Cr2O7 is mixed with KCl, NaCl precipitates out while hot because of its less solubility in comp. to KCl. When the mother liquor cooled, the orange crystals of K2Cr2O7 separates out.
Properties: It is orange-red coloured crystalline compound. It is moderately soluble in cold water but freely soluble in hot water It melts at 398°C. On heating strongly, it decomposes liberating oxygen.
Action of alkali:
On heating with alkali, it is converted into chromate i.e., the colour changes from orange to yellow. On acidifying, the yellow colour again changes back to orange.
Thus dichromate and chromate ions are inter convertible in aq. solution depending upon the pH of solution being in equilibrium at pH 4.0. The oxidation state of Cr atom is same in both – the dichromate and chromate ion.
The chromate ion is tetrahedral, the dichromate ion being to tetrahedrons sharing one corner.
Reaction with H2O2:
Acidified solution of ions develops a deep blue colour with H2O2 in presence of little ether due to formation of perchromic acid (perodie of chromium).
The oxidation state of Cr is + 6 in CrO5 as the 4 oxygen atom are present as two peroxy linkage hence in (-1) state i.e.,
x + (-2) + (-2) × 2 = 0
x = + 6
The blue colour of the solution fades away gradually due to decomposition of CrO5 into Cr3+ ions and O2.
Note: The reaction is used for the test of H2O2.
Potassium dichromate reacts with hydrochroric acid and evolves chlorine.
It acts as a powerful oxidising agent in acidic medium (dilute H2SO4).
The oxidation state of Cr changes from +6 to +3. Some typical oxidation reactions are given below
(i) Iodine is liberated from potassium iodide.
The equation in terms of electron method may also be written as
(ii) Ferrous salts are oxidised to ferric salts.
(iii) Sulphites are oxidised to sulphates.
(iv) H2S is oxidised to sulphur.
(v) SO2 is oxidised to H2SO4.
When the solution is evaporated, chrome-alum is obtained.
(vi) It oxidises ethyl alcohol to acetaldehyde and acetaldehyde to acetic acid.
It also. oxidises nitrites to nitrates, arsenites to arsenates, thiosulphite to sulphate and sulphur etc.
Chromyl chloride test: This is a test of chloride. When a mixture of a metal chloride and potassium dichromate is heated with conc. H2SO4, orange-red vapours of chromyl chloride are evolved.
When chromyl chloride vapours are passed through NaOH solution, yellow coloured solution is obtained.
Note: It is the confirmatory test of Cl– ion. All metal chlorides (except Hg, Ag, Pb and Sn) respond to this test.
Uses:
i) As oxidizing agent in laboratory.
ii) For volumetric estimation of reducing agents like oxalic acid, Fe2+, salts I– SO32– ions etc.
iii) For prep. of other chromium compounds eq. chrome alum, chrome yellow, chrome red, zinc yellow etc.
iv) In photography for hardening of gelatin film.
v) In dying by mordant dye for providing Cr(OH)3 as more mordant are those substances which have more than one group attached with it and are capable of forming bonds with dyes as well as the object to be dyed. In fact they react firstly with the fabric or leather forming a layer thought. This layer, now depending on its chemical nature be stained (dyed) with suitable colouring materials. Use of mordant become crucial for the reason that some dyes are not able enough to bound with fabric or leather permanently. As they are exposed to sunlight, acids or alkalis, their colours fade away. Use of mordant makes them acid, base or u v fast.
vi) Chromic acid (K2Cr2O7 + H2SO4) is used as cleansing agent for glass ware in lab.
vii) IN chromyl chloride test.
viii) In deflection of H2O2.
Note: While K2Cr2O7 is used in laboratory as primary standard in volumetric analysis (titration) because of it can be weighed accurately Na2Cr2O7 because of being hygroscopic can’t be used in place of K2Cr2O7.
Potassium Permanganate, (KMnO4):
This is the most important and well known salt of permanganic acid. It is prepared from the pyrolusite ore.
Preparation:
i) Disproportination of Potassium Manganate (K2MnO4):
Manganate ion dispropoertionates into manganate ion and manganese dioxide in acidic medium.
ii) Oxidation of K2MnO4 by Cr2 or O3
iii) Commercial production:
Alkaline oxidative fusion of pyrolusite (MnO2) followed by the electrolytic oxidation of the manganate ion so formed.
The pyrolucite is first fusel with KOH in presence of some oxidizing agent (KNO3 or KClO3) or atmospheric oxygen in a muffle furnace to give K2NMnO4 as green mass. The resulting mass is reached with water and the solution is oxidized to KMnO4 either by Cl2, O3 or CO2 as above or by electrolytic oxidation.
At anode (oxidation)
At cathode (reduction)
Passing CO2 through solution of K2MnO4 also results into the disproportionation and KMnO4 is produced:
The electrolysis is carried out in as non diaphragm cell, having an iron vessel with iron rods as cathode and sheet of iron as anode. A high current density is passed to give ions at anode and H2 + cathode.
Properties: It is purple coloured crystalline compound. It is fairly soluble in water. When heated alone or with an alkali, it decomposes evolving oxygen.
On treatment with conc. H2SO4, it forms manganese heptoxide via permanganyl sulphate which decomposes explosively on heating.
Potassium permanganate is a powerful oxidising agent. A mixture of sulphur, charcoal and KMnO4 forms an explosive powder. A mixture of oxalic acid and KMnO4 catches fire spontaneous after a few seconds. The same thing happens when glycerine is poured over powdered KMnO4.
Potassium permanganate acts as an oxidising agent in alkaline, neutral or acidic solutions.
(a) In alkaline solution: KMnO4 is first reduces to manganate then to insoluble manganese dioxide. Colour changes first from purple to green and finally becomes colourless. However, brownish precipitate is formed.
(b) In neutral solution: MnO2 is formed. Brownish ppt. is observed.
(c) In acidic solution (in presence of dilute H2SO4):
Manganous sulphate is formed. The solution becomes colourless.
2KMnO4 + 3H2SO4 → K2SO4 + 2MnSO4 + 3H2O + 5 [O]
or
or
This medium is used in quantitative (volumetric) estimations. The equivalent mass of KMnO4 in acidic medium is . The oxidation reactions of acidified KMnO4 are catalysed by Mn (II) ion.
The important oxidation reactions are:
In Acidic Medium:
(i) Ferrous salts are oxidised to ferric salts.
(ii) Potassium iodide is converted to iodine.
(iii) H2S is oxidised to sulphur.
(iv) SO2 is oxidised to H2SO4.
2KMnO4 + 5SO2 + 2H2O → K2SO4 + 2MnSO4 + 2 H2SO4
(v) Nitrites are oxidised to nitrates.
2KMnO4 + 5KNO2 + 3H2SO4 → K2SO4 + 2MnSO4 + 5KNO3+ 3H20
(vi) Oxalic acid is oxidised to CO2.
(vii) Hydrogen halides (HCI, HBr or HI) are oxidized into X2(halogen).
2KMnO4 + 3H2SO4 + 10HX → K2SO4 + 2MnSO4 + 8H2O + 5X2
In Neutral Medium:
(i) H2S is oxidised to sulphur.
(ii) Manganese sulphate is oxidised to MnO2
2KMnO4 + H2O → 2MnO2 + 2KOH + 3[O]
[MnSO4 + H2O + [O] → MnO2 + H2SO4] x 3
2KOH + H2SO4 → K2SO4 + 2H2O
2KMnO4 + 3MnSO4 + 2H2O → K2SO4 + 5MnO2 + 2H2SO4
(iii) Sodium thiosuiphate is oxidised to sulphate and sulphur.
2KMnO4 + 3Na2S2O3 + H2O → 2KOH + 2MnO2 + 3Na2SO4 + 3S
In Alkaline Medium
(i) Iodide is oxidized to iodate.
2KMnO4 + H2O → 2KOH + 2MnO2 + 3[O]
KI + 3[O] → K103
2KMnO4 + KI + H2O → 2KOH + 2MnO2 + KIO3
(ii) Ethylene is oxidized to ethylene glycol.
In alkaline medium it is called Baeyer’s reagent.
Illustration 10: Why is it not advisable to dissolve KMnO4 in conc. H2SO4?
Solution: With conc. H2SO4, KMnO4 reacts to form Mn2O7 which on warming decomposes to MnO2.
Uses:
i) Oxidizing agent in laboratory & industry.
ii) in volumetric analysis – as intermediate solution generally.
iii) As Baner’s reagent in organic chemistry for test of unsaturation the products being colourless, the decolourisation of pink colour of the reagent takes place.
iv) In qualitative analysis for detection of a halides sulphites, oxalates etc.
v) Bleaching of wool, cotton, silk and other textiles.
vi) Decolourization of oils
vii) In dry cells
viii) In dry cells for water – as being oxidant kills bacteria and doesn’t alter the taste of it unlike Cl2 which although disinfect but has an pleasant test.
KMnO4 is used as an intermediate solution in the titrations e.g.., quantitative estimation of Mn, iodine, thiosulphate etc. But it cannot be used as primary standard, because of following reason:
i) difficulty in obtaining pure samples, i.e., free from traces of MnO2.
ii) distilled water has some traces of reducing agents (organic matter etc) which react with some to give MnO2 and changes it’s the actual concentration.
iii) In acidic pH and in presence of sunlight it decomposes into MnO2 and hence is always stored in dark coloured bottles, away from sunlight.
Hence, it is always used as second standard solution by titrating (standardizing) it against a primary standard e.g., oxalic acid.
A primary standard is one which has definite and stable structured formula as well as its solution, which does not undergo any practical change under normal atmospheric conditions, while storing.
Copper:
Copper has been known to mankind from prehistoric times. It was alloyed with other metals and the use of bronze in Egypt is reported as early as 350 B.C. Romans and Greeks obtained this metal from the island of Cyprus from which the name ‘cuprum’ was derived.
Occurrence: Copper constitutes only 0.0001 per cent of the earth’s crust. Its deposits, however, are concentrated. Copper is found in nature in the following forms.
1. Native state: Copper is found in the metallic condition in large quantities near lake Superior in U.S.A.; in the Ural mountains (Russia) and Sweden.
2. Combined state: The principal ores of copper are sulphides, oxides and basic carbonates.
Sulphide Ores:
(i) Chalcopyrites or copper pyrites, CuFeS2
(ii) Chalcocite or copper glance, Cu2S
(iii) Bornite, Cu3FeS3
Oxide Ore
Cuprite (red), Cu2O
Basic Carbonates:
(i) Malachite (green), CuCO3.Cu(OH)2
(ii) Azurite (blue), 2CuCO3.Cu(OH)2
Copper is found to a very minute extent in the animal body where it is said to catalyse the action of iron in the formation of haemoglobin. It is present in traces in the cereals and potatoes. It occurs as a red colouring matter of the feathers of certain birds. Milk has a very low copper content.
India is not rich in copper ores. Copper is found in India mainly in Singhbhum district (Bihar), Matigara and Dharwar. The copper belt in Rajasthan (Khetri) is at present under extensive development.
Extraction:
Copper may be extracted by different methods depending upon the nature of the ore and the % of copper. The methods may be divided into two categories.
1. From sulphide ores : (a) Dry process or smelting process : This is applied to those ores in which copper content is more than 3%.
(b) Wet process or hydro-metallurgical process: This is applied to poor ores containing small percentage of copper.
2. From non-sulphide ores: Leaching process.
Dry process for the extraction of copper: Copper is extracted mainly from copper pyrites by dry process or smelting process. The process of extraction of copper from copper pyrites involves the following steps:
(i) Concentration of the ore: The concentration of the sulphide ores is done by froth floatation process. The ores are powdered and sieved and then thrown into tanks of water to which pine oil and potassium xanthate have been added. A strong stream of air is passed which agitates the whole mass. Froth is produced which carries along the particles of the ore to the surface of the liquid while the impurities settle to the bottom of the tank (Fig.). The froth is continuously separated. This is the concentrated ore.
(ii) Roasting: The concentrated ore is heated strongly in a current of air on the hearth of the reverberatory furnace (Fig). During roasting the following changes take place.
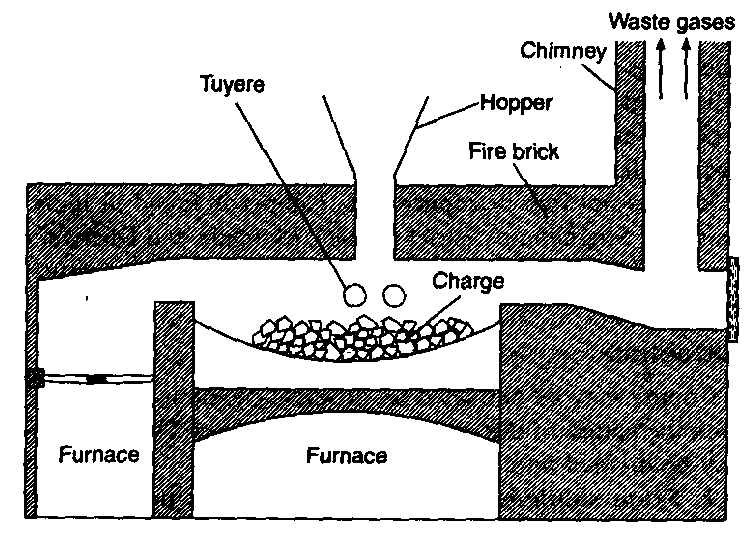
(a) Free sulphur is oxidised and removed as sulphur dioxide.
S + O2 → SO2
(b) The arsenic and antimony present in the ore are removed as volatile oxides.
4As + 3O2 → 2As2O3
4Sb + 3O2 → 2Sb2O3
(c) The pyrite is converted into cuprous sulphide and ferrous sulphide with evolution of sulphur dioxide.
2CuFeS2 + O2 → Cu2S + 2FeS + SO2
(d) The sulphides of copper and iron are partially oxidised.
2FeS + 3O2 → 2FeO + 2SO2
2Cu2S + 3O2 → 2Cu2O + 2SO2
(iii) Smelting: The roasted ore is mixed with coke and silica and transferred to a small blast furnace. The mixture is heated in the presence of excess of air. The modern blast furnace is made of steel lined inside with refractory bricks and is about 15 to 20 feet in height. It is water jacketed throughout and is provided near the top with a waste gas outlet.
The air blast enters the furnace through tuyeres following changes occur in the blast furnace.
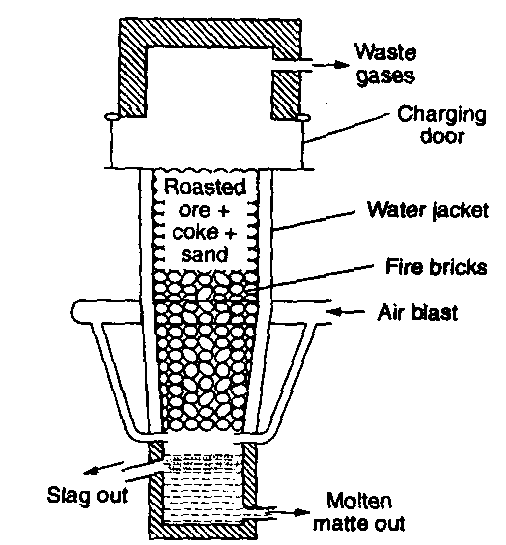
(a) The cuprous oxide reacts with ferrous sulphide.
FeS + Cu2O → FeO + Cu2S
Fe has greater affinity for oxygen than copper. The copper oxide formed reacts with unchanged iron sulphide to form iron oxide and reproduce copper sulphide. So it is difficult to oxidise cuprous sulphide until whole of the iron sulphide is oxidised.
(b) Most of the iron sulphide is oxidised to ferrous oxide.
2FeS + 3O2 2FeO + 2SO2
(c) Ferrous oxide combines with silica and forms ferrous silicate. By this reaction most of the iron is removed as slag.
The lowest point of the furnace consists of a shallow hearth in which the molten mass collects which is known as ‘Matte’. The matte contains mostly cuprous sulphide with a little iron sulphide.
(iv) Bessemerisation: The matte obtained from smelting is transferred to a Bessemer converter. Some sand (silica) is added and a blast of air is blown through the molten mass. The Bessemer converter is usually a pear shaped steel vessel lined with magnesite and quartz. It is fitted with air-blast tuyeres and mounted in such a way that it can be tilted in the desired direction (Fig. 14.10).
The following reactions occur in the Bessemer converter.
(a) Remaining ferrous sulphide gets oxidised.
2FeS + 3O2 → 2FeO + 2SO2
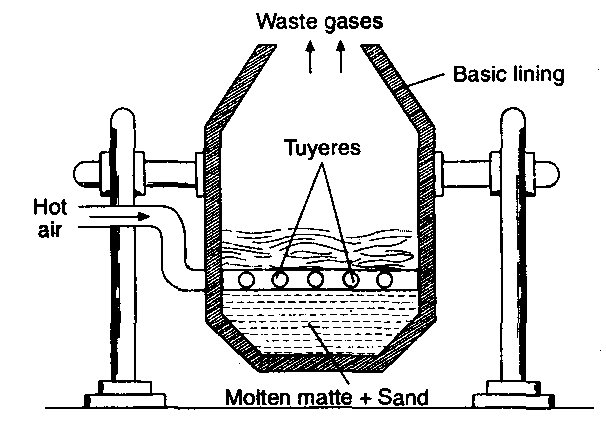
(b) Ferrous oxide combines with silica to form slag which is drained out at intervals by tilting the vessel. In about three hours, all the iron is removed as ferrous silicate.
(c) The blast of air is continued for almost another two hours. Excess of silica is absorbed by basic lining of the converter and part of cuprous sulphide is oxidised which combines with remaining cuprous sulphide to form free copper metal.
This presents an example of auto-reduction.
The molten copper is poured off and allowed to cool. During cooling, the dissolved sulphur dioxide comes out and large blisters- are formed on the surface. Hence, the metal formed is given the name ‘blister copper’. Blister copper consists 98% copper and 2% impurities.
(v) Refining: Blister copper is subjected to refining by either of the following two methods.
(a) Refining by poling: The impure metal is melted in a reverberatory furnace lined with silica. A part of copper metal is oxidised to cuprous oxide which dissolves in the melt and supplies oxygen to the more basic elements contained in it as impurity. These oxides either volatilise or combine with silica forming slag. The oxide of copper which remains in the mass is reduced by introducing poles of green wood. The gas (hydrocarbons) bubbles originating from the wood act as reducing agents. The mass is stirred vigorously with these poles. The process is thus called poling.
This process produces copper of about 99.5% purity and is known as tough pitch copper.
(b) Electrolytic refining: Copper is usually refined electrolytically. The electrolytic bath consists about 15% copper sulphate solution and 5% sulphuric acid. The anodes are of blister copper and cathodes are thin sheets of pure copper (the following fig).
As the current flows, copper from anodes dissolves while pure copper is deposited on the cathodes.
The more electropositive impurities like Fe, Zn, Ni, Co, etc., dissolve in the solution and less electropositive impurities such as Ag, Au and Pt collect below the anode in the form of anodic mud or sludge. Electrolytic copper has a purity of 99.96 – 99%.
Wet process or Hydrometallurgical process:
The essential principle of this process consists in the conversion of copper present in the ore into soluble copper compound and precipitation of copper by addition of iron or by electrolytic process.
Big heaps of copper sulphide ores are exposed to air and rain. Over a period of time, the copper sulphide is slowly oxidised to copper sulphate. The liquor which flows from the bottom of the heaps is run into pans. Copper is precipitated from the liquor by the addition of scrap iron. The precipitate is dried, melted and refined.
2. Extraction of copper from non-sulphide ores:
The oxide and carbonate ores are crushed and concentrated by gravity process. The concentrated ores are calcined in reverberatory furnace. The carbonate decomposes to form the oxide and the impurities either volatilise or are oxidised.
CuCO3.Cu(OH)2 → 2CuO + CO2 + H2O
The oxide so formed is either reduced with carbon in reverberatory furnace or leached with dilute H2SO4
CuO + C → Cu + CO
Cu2O + C → 2Cu + CO
or CuO + H2SO4 → CuSO4 + H2O

Copper sulphate solution obtained is then electrolysed using copper sheet as cathode and lead plate as anode. Copper can also be recovered by using scrap iron.
CuSO4 + Fe → Cu + FeSO4
Properties:
(a) It is a reddish coloured lustrous metal.
(b) It is highly malleable and ductile.
(c) It has specific gravity 8.9.
(d) It has high melting and boiling points (m.pt. 1083°C, b.pt. 2580°C).
(e) It is good conductor of heat and electricity (next to silver).
Silver:
Occurrence: Silver is a rare element. However, it was known in prehistoric times and used for its colour and beauty as a precious metal. Silver is found in nature in two forms.
1. Native state: It occurs in the metallic condition usually associated with copper, gold and platinum metals. Native silver has been reported in a few places in Canada, United States of America, Mexico and Peru.
2. Combined state: The important ores of silver are:
(a) In the form of sulphide:
(i) Argentite or silver glance, Ag
(ii) Pyrargyrite or Ruby silver, 3Ag
(iii) Stromeyerite or silver copper glance, (Cu, Ag)
(iv) Silver is also associated in the form of Ag in the lead ore, galena (PbS). The lead extracted usually contains silver and called argentiferous lead. Silver is recovered before lead is put into use.
(b) In the form of halide:
Chlorargyrite or Horn silver, AgCI.
In India, it occurs mainly as Ag associated with lead and zinc ores at Bawdivin and also associated with gold, in the native gold ores of Kolar. From these, silver is obtained as a by-product but no workable deposits of silver are found in India.
The ores of silver are usually associated with large amounts of rock, silica and clay. The silver content hardly exceeds 1.0 per cent. Most of the silver (nearly four-fifth of the total production) is obtained from argentiferous lead and from the anodic mud formed during electrolytic refining of copper. Thus, silver is obtained from the following sources.
(i) From the ores of silver (ii) From native silver
(iii) From argentiferous lead (iv) From the anodic mud in copper refining.
1. Extraction of silver from argentite ore (Cyanide process):
Cyanide process is the modern process for the extraction of silver. The process is also called as Mac-Arthur and Forest process. It is based on two points.
(i) Silver compounds (or even free silver) dissolve in sodium cyanide solution forming a complex salt, NaAg(CN)2 in presence of air.
(ii) Silver is precipitated from this complex salt by the addition of zinc.
The process involves the following steps:
(i) Concentration of the ore: Concentration of the ore is done by froth floatation process. The ore is crushed and taken in a tank filled with water to which pine oil and potassium xanthate have been added. The whole mixture is agitated by passing a strong stream of air. The ore particles come to the surface along with froth while silicious impurities settle to the bottom.
(ii) Cyanidation: The concentrated ore is ground to a very fine powder in ball mill The finely powdered ore is treated with dilute solution (0.4 to 0.6%) of sodium cyanide and a current of air is blown through the whole mass. The silver present in the ore dissolves in the solution slowly to form sodium argentocyanide.
The above reaction is reversible. The air which is blown in serves an important function in removing sodium sulphide from the equilibrium mixture and causing the reaction to proceed in the desired direction.
The soluble sodium argentocyanide is removed by filtration. Metallic silver and silver chloride (Horn silver) also dissolve in sodium cyanide solution.
(iii) Recovery of silver: Silver is precipitated from the solution by addition of zinc powder in a finely divided condition. Silver is precipitated as a dark amorphous mass while zinc goes into the complex.
Zn is more electropositive than silver. The precipitated silver is removed by filtration, dried and fused with potassium nitrate in a crucible in furnace. The impurities are oxidised and rise as a scum on the surface. Liquid silver on cooling appears as compact mass.
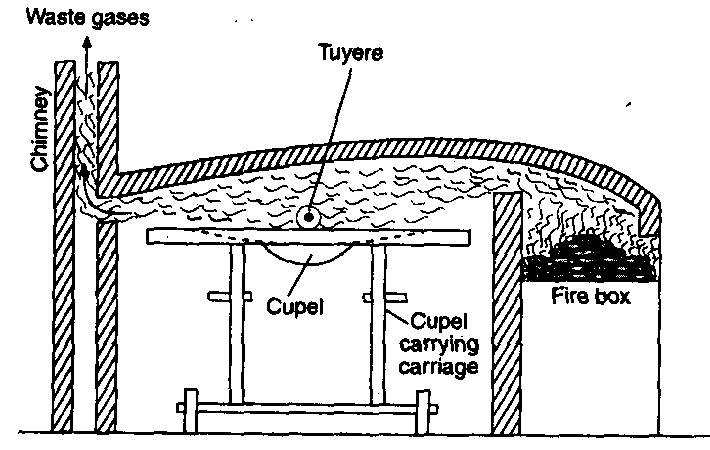 (iv) Refining: The main impurities are of lead, copper and gold. These are removed by following methods.
(iv) Refining: The main impurities are of lead, copper and gold. These are removed by following methods.
(a) Cupellation process: Cupel is a big oval dish with a shallow hearth and is made of bone-ash or porous cement. The impure silver is fused on the hearth of the cupellation furnace and a strong current of air is blown over it.
Lead is oxidised to lead oxide (litharge: PbO) which is blown away by air. Other impurities are also oxidised and rise to the surface and removed as scum. The oxides may also be absorbed by the lining of the cupel. The completion of the process is indicated by the appearance of bright shining surface of the molten silver.
(b) Electrolytic process: The impure silver is made as anode and pure silver plate as cathode. The electrolytic solution is of silver nitrate containing 10% nitric acid. On passing electric current silver ions start depositing on cathode and equivalent amount from anode comes into solution. In this way silver is transferred from anode to cathode. Copper goes into solution as copper nitrate while gold collects below the anode as anodic mud.
2. Amalgamation process: This is the old method of extraction. It is still in use in some countries. The sulphide ore is crushed and converted into a slime with a solution of cupric chloride.
Ag + CuCl → 2AgCI + CuS
Some mercury is then added to the product. Silver chloride reacts with mercury liberating silver.
2AgCI + 2Hg →2Ag + Hg2Cl2

The silver dissolves in excess of mercury to form an amalgam. It is washed and then distilled when silver is left behind in the retorts.
3. Extraction of silver from argentiferous lead (Desilverisation of lead):
Lead ores, especially galena, contain a very small percentage of silver sulphide. During the extraction of lead, silver remains in the metal. On account of its high value, silver is removed from the lead before it is used for any purpose. The crude lead contains upto 2% of silver. This poor amount of silver requires to be concentrated before desilverisation. The recovery of silver from crude lead (argentiferous lead), thus involves two steps:
(i) Partial separation of lead
(a) by Pattinson’s process or (b) by Parke process.
(ii) Removal of lead as lead oxide by cupellation process.
(a) Pattinson’s process: This process is based on the fact that silver-lead system has an eutectic mixture with 2.6% silver melting at 303°C whereas pure lead melts at 327°C.
When the molten argentiferous lead is allowed to cool slowly, crystals of pure lead are deposited until the silver content of the mixture has risen to 2.6%. The crystals of pure lead are removed with the help of perforated ladles. The alloy rich in silver content is then subjected to cupellation to remove the remaining lead.
(b) Parke process: This is at present the most commonly used method. It depends on the following points:
(i) Zinc and lead are not miscible.
(ii) Silver is more miscible with zinc than lead.
(iii) Zinc-silver alloy is lighter than molten lead and have a higher melting point.
Lead containing silver is melted in large pots. Zinc dust to the extent of only 1% is added and thoroughly stirred. The temperature is raised above the melting point of zinc. Zinc dissolves silver and comes up on the surface where zinc-silver alloy forms a crust. This is skimmed off by perforated ladles. More zinc is added and the operation is repeated for several times till lead shows almost no trace of dissolved silver. It is possible to reduce the silver content to 0.0005%.
The zinc-silver alloy containing some lead is now distilled with a little charcoal when zinc distills over. The recovered zinc is used again. The lead silver alloy left behind is now put to cupellation as to remove lead. The pure silver is, thus, obtained.
4. Recovery of silver from anodic mud of copper refining: The mud is treated with nitric acid and the solution is filtered. The filtrate contains nitrates of silver and lead. To the filtrate HCI is then added when silver and lead settle down as chlorides in the form of white precipitate. The chlorides are separated, dried and fused with sodium carbonate in a crucible when an alloy of Ag and Pb is obtained. To remove lead, the alloy is put to cupellation.
5. Sliver from coins or ornaments: Coins and ornaments are alloys of silver and copper. The alloy is treated with nitric acid. Both copper and silver go into the solution in the form of nitrates. The excess of nitric acid is boiled and the solution is treated with dil. HCl when a white precipitate of silver chloride is obtained. This is separated and converted into silver by any of the following methods.
(a) Silver chloride is fused with sodium carbonate in a crucible.
(b) The precipitate of AgCl is reduced with nascent hydrogen produced by the action of zinc and dilute H2SO4.
Zn + H2SO4 → ZnSO4 + 2H
AgCl + H→ Ag+HCl
(c) AgCI is dissolved in potassium cyanide solution when a complex alt, potassium argento-cyanide is formed.
AgCl + 2KCN → KAg (CN)2 + KCl
Ag is obtained from the solution by addition of zinc.
(d) By boiling silver chloride with caustic soda and glucose.
The silver thus obtained, is purified by fusion with borax and nitre followed by electrolytic purification.
Separation of silver from gold: The separation of silver and gold is technically known as parting. This is done by following methods:
(a) Parting with sulphuric acid: The alloy having gold, less than 20%, is treated with boiling concentrated sulphuric acid. Silver dissolves as silver sulphate and gold remains as spongy mass. The solution is diluted with water and the solution is filtered. The filtrate is then treated with scrap iron or copper or zinc to separate silver.
Ag2SO4 + Zn → 2Ag + ZnSO4
If the percentage of gold in the alloy is more than 20%. some silver is added to alloy as to reduce the percentage of gold less than 20% and then the process of parting is applied.
(b) Parting with ‘Moebius’ electrolytic process: The alloy of silver and gold is made the anode while pure silver plate as cathode. The electrolytic solution consists of a dilute solution of silver nitrate acidified with nitric acid. On passing electric current silver dissolves from the anode and deposits on the cathode. Gold remains undissolved and deposits as a slime in canvas bags surrounding the anode.
Properties:
Physical: (a) It is a lustrous white metal. It melts at 961°C and boils at 2180°C. Its specific gravity is 10.5. (b) It is very good conductor of heat and electricity (better than even copper). (c) On heating, the molten mass absorbs oxygen which is again released on cooling. This property is called spitting of silver. (d) It is hard, malleable and ductile.
Chemical:
(a) Action of atmosphere: Silver remains untarnished in air free from hydrogen sulphide Air contaminated with hydrogen sulphide covers it with an adherent film of black silver sulphide.
2Ag + H2S →Ag2S + H2
(b) Action of oxygen and water: Silver is hot affected by oxygen and water.
(c) Action of halogens: Silver combines slowly with free halogens even at room temperature. The reaction is rapid at red heat.
2Ag + X2 →2AgX (X2 = Cl2, Br2 or I2)
(d) Action of acids: Silver is not affected by dilute and concentrated hydrochloric acid. It is also not acted upon by dilute sulphuric acid. In concentrated sulphuric acid, silver dissolves on heating with evolution of sulphur dioxide.
Silver dissolves in both dilute and concentrated nitric acids.
Dilute HNO3 : 2HNO3 → H2O + 2NO + 3[O]
or
Conc. HNO3 :
or
(e) Action of alkalies: Alkalies have no action on silver even when fused.
(f) Action of alkali cyanides: Silver dissolves in the solution of alkali cyanides in the presence of oxygen forming the complex argentocyanide.
Uses of silver:
i) Making coins, jewellary and for decorative purpose.
ii) Silver plating of metallic articles e.g., table wares.
iii) Reflexive layers of good quality silver mirror.
iv) Photography (major use of Ag as Ag Br)
v) In filling silver amalagam.
vi) Pure silver is beaten into thin leaves used in Ayurvedic and Unani medicines as tonic.
vii) High capacity batteries
viii) Disposing off the stock pile of chemical weapons.
Silver is used in making coins, ornaments, silver ware, decoration pieces, etc. Pure silver is too soft to be used for these purposes and is, therefore, alloyed generally with copper. Silver ornaments and utensils usually contain 80% silver and 20% copper. A silver coin has generally the composition Ag 50%, Cu 40%, Zn 5% and Ni 5%.
(ii) Silver is used for plating articles of base-metals. The article to be plated is made the cathode while the anode consists of pure silver. The electrolyte is the solution of potassium argentocyanide. As the complex ion is very stable, the concentration of silver ion in solution is very small at any time.
Silver is deposited slowly and uniformly when electricity is passed through the solution.
Halides:
The transition metals react with halogens at higher temperature and form a variety of halides. The reactivity of halogens:
While reacting with F2, the metals are generally oxidized to their highest oxidation states, while the lower oxidation states are stabilized in the iodides. The nature of ionic bond decreases from fluorides)
Silver halides:
All the silver (I) halides are known. The fluoride (AgF) is unique in being water soluble whereas the chlorides, bromides & iodides are insoluble in water.
AF is prepared by the action of HF on Ag2O and crystallizing the solution.
The chloride, bromide and iodide are made mostly by addition of halide ion (X–) to the solution of silver ion (Ag+ e.g., AgNO3). The colour and insolubility increase in the order of Cl < Br < I (from white to yellow).
The chloride and bromides and soluble in NH3 solution (i.e., NH4OH) with the formation of soluble linear complex [Ag(NH3)2]+. Cl– being readily soluble while Br– sparingly. I– remains insoluble.
Silver halides are light sensitive i.e., darken upon exposure to light due to photochemical decomposition into metallic Ag, black in colour. Hence used in photography.
All silver halides are soluble in thiosulphate and cyanide solutions forming dithiosulphato and dicyanato complexes.
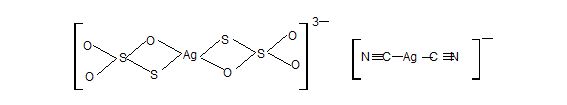
Uses of silver halides:
AgCl – Photography – Chiefly for printing paper & lautern slides.
AgBr – Photography – Photographic plates and filens
AgI – Photography – Colloidal emulsion plates.
Illustration 11: Why is AgBr used in photography?
Solution: Out of all silver halides, AgBr is the most sensitive to light and undergoes photo-reduction to metallic silver instantaneously on exposure to light.
Photography:
The art of obtaining an exact impression of an object on a plate or paper by a chemical reaction initiated by light is called photography. It is based on the nature of silver halides. Except AgF, the silver halides are photosensitive. Silver. halides, particularly silver bromide, undergo decomposition in light and turn black due to formation of free silver.
The entire process of photography involves the following steps:
1. Preparation of photographic plate or film: An emulsion of silver bromide is prepared in a dark room by mixing silver nitrate solution with ammonium bromide solution containing gelatine with constant stirring.
NH4Br + AgNO3 → AgBr + NH4NO3
The emulsion is allowed to stand at about 45°C for some time so that the particles of silver bromide may grow in site. This treatment is termed as the ripening of emulsion. The emulsion is now cooled in ice as to solid it is washed with water in order to make it free from ammonium nitrate. It is warmed to melt it again.
The melted emulsion is applied uniformly on a celluloid film or smooth glass plate. Whole of this operation should be done in a dark and dust-free room.
2. Exposure: The light-sensitive film or plate is loaded in a camera which is focused on the object to be photographed. When the shutter of the camera is opened for a few seconds, the light from the object through the lens falls on the film or plate. The silver bromide is affected by light and gets activated. This is not a visible change but is called formation of latent image. The light reduces silver bromide. The effect produced on the film or plate is directly proportional to the intensity of light.
2AgBr →2Ag + Br
Bromine is absorbed by gelatine and helps in the decomposition of silver bromide. According to one theory (Gurney-mon theory), the following sequence of events occur:
(i) An incoming quantum of light hu kicks an electron out of Br– to form Br and e–.
(ii) The electron wanders through the crystal of AgBr and eventually gets trapped at a surface defect.
(iii) An interstitial Ag+ , such as is commonly found in AgBr, diffuses to the trap site where the Ag and the trapped eT combine to give Ag.
(iv) A second quantum of light energy hu comes along and ejects a second electron which migrates to Ag and converts it into Ag.
(v) A second interstitial Ag subsequently diffuses over and combines.
Ag+ + Ag– → Ag2
(vi) The process repeats until a clump of about 50 silver atoms is built up. The AgBr grain is now “activated”.
3. Developing: The treatment of the exposed photographic film with reducing agents is called developing of the film. The chemicals used for developing as reducing agents are called developers. A developer is usually a weak reducing agent such as potassium ferrous oxalate or an alkaline solution of pyrogallol or an alkaline solution of quinol, etc. The exposed film or plate is kept in a solution of a developer for some time. The parts activated by light are reduced to deposit more of black silver.
The developer does not affect those parts of the photographic film or plate which are not activated by light. On the developed film or plate the shades of the object are reversed, Le., the bright parts of the object appear dark and the dark parts appear bright. The plate or film is, therefore, called a negative. This operation is carried out in dark.
4. Fixing: In order to make the image permanent, it is necessary to remove the unreduced silver bromide from the surface of the developed film. This operation is called fixing of image. Fixing is done by dipping the developed film or plate in sodium thiosu (hypo) solution. The hypo solution dissolves the unreduced silver bromide by forming a complex.
This operation is also done in dark. The plate is now thoroughly washed with water and dried. It can now be taken out of the dark room into light.
5. Printing: To get a positive image, the whole process is repeated. The Printing Out Paper (POP) or bromide paper is used. The POP has got a coating of silver bromide emulsion. The negative is placed over POP and then exposed to light for a fraction of a second. A negative of the negative plate or positive with respect to the actual object is obtained on the print paper. The print paper is then subjected to developing and fixing as usual. The positive print has shades exactly similar to those of the object.
6. Toning: It is a technique by which different shades can be given to the printed photograph with the help of chemicals. The printed photograph obtained is black and white. This photograph is dipped in a solution containing the salt solution of either gold or platinum or selenium, etc. The dark silver particles are replaced by another metal. Thus, gold salt gives golden shade and platinum salt makes the shade bright grey.
AuCl3 + 3Ag → + 3AgCI
PtCI4 + 4Ag →
The various steps involved in photography are shown in figure.
Zinc:
Occurrence: Zinc is usually found in the combined state although traces of the metal in the native state have been reported from Melbourne (Australia). Its chief ores are:
1. Zinc blende, ZnS. It is found in Burma, Belgium, Silesia and Oklahoma.
2. Calamine or zincspar, ZnCO3
3. Zincite, ZnO
4. Willemite, Zn2SiO4
Traces of zinc in the form of organo-metallic compounds have been reported in the animal cells and in snake-poison. In India, zinc is mainly found at Zawar in Rajasthan.
Extraction: Zinc is extracted from its ores by two methods:
1. Reduction process and
2. Electrolytic process.
Reduction process: It involves the following steps:
(i) Concentration: When zinc blende is used, the powdered ore is concentrated by froth floatation process. In the case of calamine ore, the concentration is done by gravity process. If the ore contains iron oxide, the latter is removed by magnetic separation.
(ii) Roasting: The concentrated ore is heated in excess of oxygen at about 900°C. Zinc sulphide is oxidised to zinc oxide. If some of the ore is oxidised to zinc sulphate, it also decomposes at 900°C into ZnO.
When the ore is calamine, it shall decompose into oxide with evolution of carbon dioxide.
ZnCO3 → ZnO + CO2
For roasting, a reverberatory furnace may be used.
(iii) Reduction: The principal reaction that takes place during reduction is the conversion of the oxide into the metal with the help of carbon.
ZnO + C → Zn + CO
There are different types of reducing furnaces for the reduction of the oxide. All make use of the same principle but differ only in the details of construction and method by which the metal is separated.
(a) Belgian process: It is an old process. The roasted ore is heated with coke to about 1100°C in a small fireclay retorts. The heating is often done by producer gas. Each retort is filled with a mixture of 2 parts ore and one part coke. The retorts are about 1.5 metre long and 25 cm in diameter. Each retort
is circular or elliptical in shape and closed at one end. The mouth of each retort is provided with an earthenware condenser. The temperature is raised to about 1100°C, the CO formed during the process burns at the mouth of the retorts and when the colour of the flame changes from blue to greenish white, the condensers are fitted there on. These condensers are cooled by air and the metal condenses in these partly as a fused metal (spelter) and partly as a powder (zinc dust).
Reactions: ZnO + C → Zn + CO
ZnO + CO → Zn + CO2
CO2 + C → 2CO
There are number of such retorts which are arranged in three or four rows one above the other.
(b) Vertical retorts process: This is a continuous process and is largely used these days. It is economical also.
In this process the vertical retorts of about 7.5 metre height are used. Each is connected with an arrangement for continuous feeding of roasted ore and powdered coke. Retorts are made
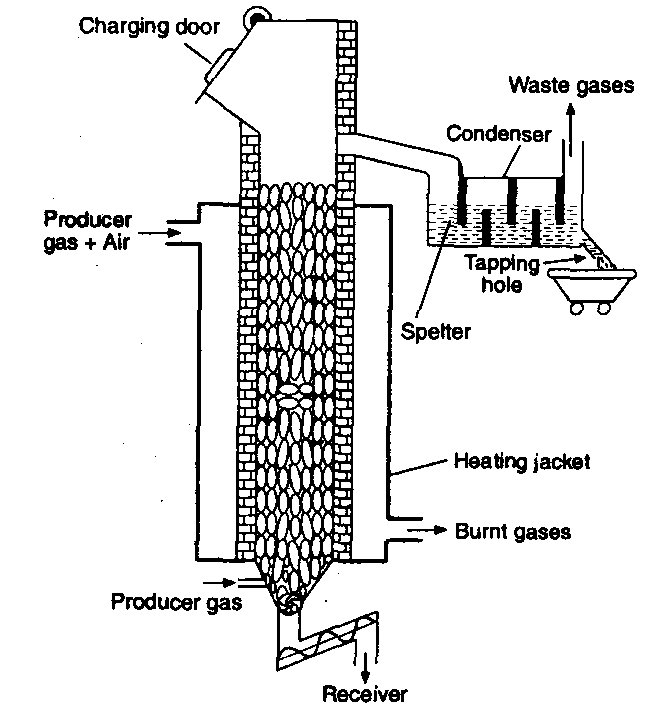
of highly refractory silicon carbide bricks capable of with standing high temperature 1300°C. The retorts are heated externally by producer gas. There is an extension at the bottom through which ash can be removed. The open end of the retort is connected with condensers. Zinc vapour and carbon monoxide pass into the condensers when zinc liquefies. Molten zinc is periodically taped off from the condensers and carbon monoxide is used as fuel for heating the furnace.
(iv) Purification: The zinc obtained above is impure containing 97.8% zinc and rest impurities of lead, iron, cadmium, arsenic, etc. This impure zinc is known as spelter. This is put to further purification by distillation. The distillation is carried around 950—1000°C when only zinc (b. pt. 907°C) and cadmium (b. Pt. 767°C) distil over. From this sample, cadmium is removed at 800°C.
The impure zinc can also be purified electrolytically. The impure metal is made anode and cathode consists of sheets of pure aluminium. A solution of zinc sulphate acts as an electrolyte. Zinc dissolves from anode and deposits on cathode when electric current is passed. Zinc is scraped off &om the aluminium sheets.
2. Electrolytic process: This method is gradually replacing the reduction process. The roasted ore is dissolved in dilute sulphuric acid and the solution is filtered. The solution is freed from iron, aluminium, silica, etc., by treatment with calcium hydroxide. Copper and cadmium are removed by precipitation with zinc dust at 75°C.
FeSO4 + Ca(OH)2 → Fe(OH)2 + CaSO4
CdSO4 + Zn → Cd + ZnSO4
CuSO4 + Zn → Cu + ZnSO4
The filtered solution is subjected to electrolysis by using a sheet of pure aluminium as cathode and pure lead plate as anode. The zinc is removed from cathode by melting. The zinc obtained is 99.95% pure.
Granulated zinc is made by melting the metal in a crucible and pouring the drops in water. Zinc sheet is formed by heating the metal at 150°C when it becomes soft and then can be rolled into sheets.
Alloys of Zinc:
|
Name |
% composition |
Use/Prop. |
|
Brass |
33%Zn 67% Cu |
Utensils etc |
|
China silver |
20% Zn 60%Cu |
Tablewares |
|
Prestal |
78% Zn 22% Al |
As strong as steel but as easy to mould as plastic. |
Uses of zinc compounds:
ZnO – as pigments in plastics, cosmetics, photocopier paper (being replaced by TiO2) and rubber industry catalyst and heat dispersing agent
ZnO – as sun blocker – against damage to UV rays – applied on lips and nose by cricketers & other sport persons or by those who are exposed to blazing sun for many hours
ZnO is better pigment than white lead as the latter gets blackened upon exposure to atmospheric H2S while the former remains unaffected
ZnO on heating with Co(NO3)2 gives a green mass of cobalt zincate known as Rinnmann’s green
ZnO + Cr2O3 – catalyst in : water gas → methyl alcohol
ZnO + Fe2O3 – Calamine lotion (now replaced by lacto calamine as face cream)
ZnS – as phosphor in X-ray screens, television & fluorescent lighting
ZnS + BaSO4 – lithopone – white paint
ZnS + 1 ppm radium salt – luminous paint for dial watches
Zn stearate – as ointment
Znl2 – as preservative for wood; dehydrating agent in org. chem.
Mercury Hg:
(Latin – hydrargyrum, liquid silver)
Mercury forms 0.1ppm of earth’s crust: Mercury occurs in small quantities in native state. In combined state, only cinnabar, HgS, is the major ore and occurs in Spain & Italy.
Extraction: Extraction of mercury from cinnabar is comparatively simple involving mainly roasting and distillation.
1. Coushing and concentration: Crushing in ball mill and concentration by froth flotation.
2. Roasting and distillation (combined): The ore is roasted in air above 3000C to convert HgS into HgO and SO2. SO2 escapes and raising the temperature upto 5000C decomposes HgO into metallic mercury.
The mercury distills over at this temperature (bp 3570C) and is condensed into pure liquid form (mp -38.870C)
The flue dust coming out from the furnace during roasting and the ore rich in HgS are mixed with lime before distillation in iron retort.
3. Purification: Commercial mercury has some oxide impurities of Zn, Cu, Pb etc and can be purified by:
i) Squeezing through linen or chamois leather to remove suspended particulate matter.
ii) More basic metals such as lead and zinc, which are easily oxidized are removed by passing dust free air through mercury at 1500 This converts these metals into scum and are skimmed off.
iii) The mercury so obtained is dropped slowly through a solution of dilo HNO3(45%) which contains a little HgNO3.
Since Hg is less active than other metals, it is displaced from the solution as Hg and the impurity of metals passed as ions into the solution.
Zn + 2HgNO3 → Zn(NO3)2 + 2Hg
iv) Further purification is carried out by distillation under reduced pressure.
Properties: Hg is silvery white heavy liquid (13.6 g cm–3). Hg vapours are very poisonous. Mercury forms spherical drops easily because of its high surface tension.
Mercury dissolves almost of metals in it and forms alloys called amalagams which are named after dissolved reacted metal, e.g., Au, Ag, Sn, Pb, Cu, Mg, Na, K etc. (Na-Hg → sodium amalgam).
Fe, Co and Ni do not dissolve in it to form amalgam directly.
Uses:
i) in thermometers, barometers and pressure gauge etc.
ii) in extraction of Ag and Au.
iii) manufacture of NaOH
iv) mercury lamps
v) mercury drugs
vi) manufacture of vermillion – HgS
vii) in amalgams – Na-Hg- as reducing agent
Sn-Hg – coating mirrors
Uses of mercury compounds:
• Hg is the only liquid metal at soon temperature (250C)
• Mercury tree: Addition of a little Hg into a AgNO3 solution gives a tree like growth of silver amalgam, called mercury tree.
• HgS – vermillion is used as cosmotic and in Ayurvedic medicine as Makardhwaja
• Hg(SCN)2 – for making Pharaoh’s serpents. Mixed with a little gum and made into pellets, when ignited yield a serpent like voluminous ash.
• Chlorides of Hg and Ag (that of Pb and Sn also) do not respond to chromyl chloride test.
• HgCl2 is strong poison. Its antidote is egg-white which eliminates it in the form of coagulated mass.
f-BLOCK ELEMENTS:
28 elements from atomic number 58 to 71(14 elements) and from atomic number 90 to 103 (14 elements) have been arranged in two horizontal rows below the periodic table. These elements are collectively called f-block elements as the last or differentiating electron in the atoms of these elements is accommodated on one of the seven f-orbitals of the ante-pen ultimate (next to the penultimate) energy shell. These elements have also been called inner transition elements because the ante-penultimate energy shell, Le., (n-2)f-orbitals, lie comparatively deep within the kernel (being inner to the penultimate shell).
f-Block consists of two series of elements known as Lanthanides or Lanthanons and Actinides or Actinons. The lanthanide series follows lanthanum (At. No. 57), a member of Sd-series. Similarly, actinide series comes after actinium (At. No. 89), a member of 6d-series. The 14 members of lanthanide series have been placed along with lanthanum in the third group and sixth period and similarly 14 members of the actinide series have been placed with actinium in the third group and seventh period. The justification for assigning one place to these elements has been given on the basis of their similar properties. The properties are so similar that the fifteen elements from La to Lu can be considered as equivalent to one element. The same explanation can be given in the case of actinides. In case these elements are assigned different positions in order of their increasing atomic numbers, the symmetry of the whole arrangement would be disrupted. Due to this reason, the two series of elements, i.e., lanthanides and actinides are placed at the bottom of the periodic table and constitute one block of elements, i.e., f-block. The general electronic configuration of the f-block elements is:
(a) 41-series (Lanthanides): There are fourteen elements from cerium (At. No. 58) to lutetium (At. No. 71) in this series. 4f-orbitals are gradually filled up. In the past, these elements were called rare earths. This name is not appropriate because many of the elements are not particularly rare. Promethium is artificial radioactive element.
(b) 5f-serles (Actinides): There are fourteen elements from thorium (At. No. 90) to lawrencium (At. No. 103) in this series. 5f-orbitals are gradually filled up. The members of actinium are radioactive and majority of them are not found in nature. The elements from atomic number of 93 onwards are called transuranic elements and have been discovered by synthetic methods, i.e., these are man made elements.
General characteristics of lanthanides:
The general characteristics are similar to transition metals, i.e.. d-block elements.
Electronic configuration: The energies of 5d- and 4f-orbitals are nearly similar and thus their fillings show certain irregularities. The electronic configurations of the atoms of the lanthanides in their ground state are given in the following table. These are the most commonly accepted configurations.
|
Name of the element |
Symbol |
Atomic number |
Electronic configuration |
Oxidation states |
|
Lanthanum |
La |
57 |
[Xe]5d16s2 |
+3 |
|
Cerium |
Ce |
58 |
[Xe]4f15d16s2 |
+3, + 4 |
|
Praseodymium |
Pr |
59 |
[Xe]4f36s2 |
+3, (+ 4) |
|
Neodymium |
Nd |
60 |
[Xe]4f46s2 |
(+2), + 3 |
|
Promethium |
Pm |
61 |
[Xe]4f56s2 |
(+2) + 3 |
|
Samarium |
Sm |
62 |
[Xe]4f76s2 |
(+2) + 3 |
|
Europium |
Eu |
63 |
[Xe]4f76s2 |
+2, +3 |
|
Gadolinium |
Gd |
64 |
[Xe]4f96s2 |
+3, +4 |
|
Terbium |
Tb |
65 |
[Xe]4f96s2 |
+3, +4 |
|
Dysprosium |
Dy |
66 |
[Xe]4f116s2 |
+3 |
|
Holmium |
Ho |
67 |
[Xe]4f116s2 |
+3 |
|
Erbium |
Er |
68 |
[Xe]4f126s2 |
+3 |
|
Thulium |
Tm |
69 |
[Xe]4f136s2 |
(+2), +3 |
|
Ytterbium |
Yb |
70 |
[Xe]4f146s2 |
+2, +3 |
|
Lutetium |
Lu |
71 |
[Xe]4f145d16s2 |
+3 |
Illustration 12: Briefly explain why electronic configurations of lanthanides are not known with certainty.
Solution: In the lanthanoids, 4f and 5d subshells are very close in energy. The outermost 6s orbital remains filled with 2 electrons (6s2).
The electronic configuration of lanthanum is [Xe]5d16s2. It is expected that 14 elements from cerium to lutetium would be formed by adding, 1, 2, 3,…14 electrons into the 4f level. However, it is energetically favourable to move the single electron on 5d into the 4f level in most of the elements but not in the cases of Ce, Gd and Lu. In Gd and Lu besides 5d1 the 4f-orbitals are half filled or filly filled. This gives extra stability to the core. The extra stability of half filled and filly filled f-orbitals is also seen in Eu (4f76s2) and Yb(4f’146s2).
Oxidation states: The common stable oxidation state of all the lanthanides is +3. The oxidation states of +2 and +4 are also exhibited by some of the elements. These oxidation states are only stable in those cases where stable 4f0, 4f7 or 4f’14 configurations are achieved.
For example, Ce4+ (4f0) Tb4+ (4f7) Eu2+ (4f7), Yb2+ (4f14) are stable. The oxidation states shown in parentheses in the above table are less stable. +2 or +4 oxidation states tend to revert to the more stable oxidation state of +3 by loss or gain of an electron Sm2+, Eu2+ and Yb2+ ions are thus good reducing agents in solutions while Ce4+, Tb4+ ions, etc., are good oxidising agents. The compounds of lanthanides are mainly ionic in nature.
Atomic and ionic radii (Lanthanide contraction): In lanthanide series, there is a regular decrease in the atomic as well as ionic radii of trivalent ions (M3+) as the atomic number increase from cerium to lutetium. This decrease in size of atoms and ions is known as Lanthanide contraction. Although the atomic radii do show some irregularities but ionic radii decrease steadily from La to Lu. However, the decrease is very small. For example,
|
Element |
Atomic radii (pm) |
Ionic radii (pm) (M3+) |
|
La |
169 |
103 |
|
Ce |
165 |
102 |
|
Pr |
164 |
99 |
|
Nd |
164 |
98.3 |
|
Pm |
— |
97 |
|
Sm |
166 |
95.8 |
|
Eu |
185 |
94.7 |
|
Gd |
161 |
93.8 |
|
Tb |
159 |
92.3 |
|
Dy |
159 |
91.2 |
|
Ho |
158 |
90 |
|
Er |
157 |
89 |
|
Tm |
156 |
88 |
|
Yb |
170 |
86.8 |
|
Lu |
156 |
86 |
On moving from Ce to Lu, the decrease in atomic radii occurs from 165 to 156 pm, i.e., the decrease is only 9pm. Similarly, the decrease in ionic radii occurs from 102 (Ce to 86 (Lu pm, i.e.. the decrease is only 16 pm. Thus, for an increase of 14 in the atomic number, the decrease in atomic mdii or ionic radii are very small in comparison to the elements of other groups and periods.
Cause of lanthanide contraction: As we proceed from one element to the next element in the lanthanide series, the nuclear charge, i.e., atomic number increases by one unit and the addition of one electron occurs at the same time in 4f- energy shell. On account of the very diffused shapes off orbitals, the 4f electrons shield each other quite poorly from the nuclear charge. Thus, the effect of nuclear charge increase is somewhat more than the changed shielding effect. This brings the valence shell nearer to the nucleus and hence the size of atom or ion goes on decreasing as we move in the series. The sum of the successive reductions is equal to the total lanthanide contraction.
Results of lanthanide contraction: The main consequences of lanthanide contraction are the following:
(i) Similar chemical properties: Since the change in the ionic radii in the lanthanide series is veiy. prop Thus, it is very difficult to separate these elements in the pure state. However lanthanide contraction brings some differences in properties like solubility, complex ion formation, hydration, etc. These differences help the separation of lanthanide elements by fractional crystallization or ion exchange methods.
(ii) Basic strength of hydroxides: ‘As the size of the lanthanide ions decreases from Ce3+ to Lu3+ the covalent character of M-OH bond increases and hence the basic strength decreases. Thus, Ce(OH)3 is most basic while Lu(OH)3 is least basic.
(iii) Similarity of second and third transition series: In vertical columns of transition elements, there is an increase in size from first member to second member as expected but from second member to third member, there is very small change in size and sometimes sizes are same. This is due to lanthanide contraction.
|
Group number |
3 |
4 |
5 |
6 |
|
1st transition series |
Sc (144 pm) |
|
|
|
|
2nd transition series |
Y (162 pm) |
Zr (145 pm) |
NB (134 pm) |
Mo (130 pm) |
|
3rd transition series |
La(Lanthanides) (168 pm) |
Hf (144 pm) |
Ta (134 pm) |
W (130 pm) |
In each vertical column of transition elements, the elements of second and third transition series resemble each other more closely than the elements of first and second transition series on account of lanthanide contraction. The pairs of elements such as Zr-Hf, Mo-W, Nb-Ta, etc., possess almost the same properties.
Physical properties: All the lanthanides are metals. They are soft, malleable and ductile in nature. They are not good conductors of heat and electricity. They are highly dense metals and their densities are in the range of 6.77 to 9.74 g cm The densities and atomic volumes, in general, increase with increase in atomic number. But a regular trend is not observed. They have fairly high melting points. However, no definite trend is observed.
Ionisation energies: Lanthanides have fairly low ionisation energies. 1E and 1E values are quite comparable with the values of alkaline earth metals, particularly calcium. The sum of the first three ionisation energies in U moF’ for each element are given below. The values are low.
|
Ce |
Pr |
Nd |
Pm |
Sm |
Eu |
Gd |
|
3512 |
3623 |
3705 |
— |
3898 |
4033 |
3744 (kJ mol–1) |
|
Tb |
Dy |
Ho |
Er |
Tm |
Yb |
Lu |
|
3792 |
3898 |
3937 |
3908 |
4038 |
4197 |
3898 (kJ mol–1) |
Due to low values of ionisation energies, lanthanides are highly electropositive in nature. These elements react with cold and hot water to liberate hydrogen. The reactions are, however, slow with cold water but fast with hot water.
The values of standard reduction potential (E° values) increase from La to Lu. E° values become less negative in the series. The values in volts are given.
|
La |
Ce |
Pr |
Nd |
Pm |
Sm |
Eu |
Gd |
|
-2.52 |
-2.48 |
-2.46 |
-2.43 |
-2.42 |
-2.41 |
-2.40 |
-2.39(volt) |
|
Tb |
Dy |
Ho |
Er |
Tm |
Yb |
Lu |
|
|
-2.39 |
-2.35 |
-2.32 |
-2.30 |
-2.28 |
-2.27 |
-2.25 (Volt) |
|
All the lanthanides are, thus, strong reducing agents. The reducing power decreases from La to Lu.
Coloured ions: Many of the lanthanide ions are coloured in solid state as well as in solutions. The colour is due to partially filled f-orbitals which allow f-f transitions. M3+ ions having 4f°, 4f7 or 4f14 configurations are colourless.
Pair of M3+ ions having the same number of unpaired electrons in 4f-orbitals have the same colour.
|
La3+ |
Gd3+ |
Lu3+ |
Pr3+ |
Tm3+ |
Nd3+ |
Er3+ |
Sm3+ |
Dy3+ |
Eu3+ |
Tb3+ |
|
(4f0) |
(4f7) |
(4f14) |
(4f2) |
(4f12) |
(4f3) |
(4f11) |
(4f5) |
(4f9) |
(4f6) |
(4f8) |
|
Colourless |
Green |
Pink |
Yellow |
Pale Pink |
||||||
[Note: The cations namely Ce3+(4f1) and Yb3+ (4f13) are colourless in spite of the fact that these ions have one f-orbital singly occupied. These exceptions are difficult to explain].
Magnetic properties: Ions having unpaired electrons are paramagnetic while those having all the orbitals paired are diamagnetic. The lanthanide ions (M3+) except La3+ and Lu3+ are paramagnetic since they contain 1, 2,…7 unpaired electrons.
Chemical reactivity: All the lanthanides have almost similar chemical reactivity. The metals tarnish readily in air and on heating in O2 form oxides of the type M2O3. The one exception is cerium which forms CeO2 rather than Ce2O3. The oxides are ionic and basic.
The metals react with hydrogen but often require heating up to 300-400°C. The products are solids of formula MH3. The hydrides are decomposed by water and react with O2. The anhydrous halides, MX3 can be made by heating the metal and halogen or by heating the metal oxide with the appropriate ammonium halide.
The fluorides are very insoluble. The chlorides are deliquescent and soluble. At elevated temperatures, lanthanides react with N, C, S, P, As, Sb and Bi. A wide variety of oxo salts are known. The carbonates, phosphates, chromates, oxalates, etc., are largely insoluble in water while nitrates, acetates, sulphates, etc., are soluble.
Because of their similar chemical reactivities, their separation from one another is very difficult.
Illustration 13: Give reasons why chemistry of all the lanthanides is quite similar.
Solution: The change in the size of the lanthanides due to lanthanide contraction is very small as we proceed from La (Z = 57) to Z = 71). Hence, their chemical properties are similar. Moreover, their valence shell configuration remains the same because the electrons are added into the inner 4f-subshell. Hence, they show similar chemical properties.
Complex formation: The lanthanides do not have much tendency to form complexes due to low charge density because of their large size. However, the tendency to form complexes and their stability increases with the increase of atomic number.
Uses of Lanthanides:
The metals are seldom used in pure state. As lanthanides do not differ much in their physical and chemical properties, these are mostly used in the form of alloys. Some common uses of lanthanides and their compounds are given below:
(i) Misch metal (an alloy) : Misch metal is an alloy consisting lanthanide metals (94—95%), iron (5%) and traces of sulphur, carbon, silicon, calcium and aluminium. The main lanthanide metals present are cerium (about 40%), lanthanum and neodymium (about 44%). These alloys are used for making ignition devices such as tracer bullets, shells and flints for lighters.
An alloy of magnesium and about 3% misch metal is used in making jet engine parts. Cerium-magnesium alloys are used in flash light powders.
(ii) Cerium salts are used in dyeing cotton, in lead accumulators and as catalyst.
(iii) Lanthanum oxide is used for polishing glass. Neodymium and praseodymium oxides are used for making coloured glasses for goggles. CeO is used in gas mantles.
(iv) Ceric sulphate is a well known oxidising agent in volumetric analysis.
(v) Many lanthanide oxides are used as phosphor in colour TV tubes.
(vi) Various compounds of lanthanides are used as catalysts for hydrogenation, dehydrogenation, oxidation and petroleum cracking.
(vii) The compounds of lanthanides are used in making magnetic and electronic devices for their paramagnetic and ferromagnetic properties.
(viii) Neodymium oxide dissolved in selenium oxy-chloride is used these days as a powerful liquid laser.
General characteristics of actinides:
Excepting Ac, Th, Pa and U which occur in nature in uranium minerals, all the remaining actinides are unstable and synthetic elements. These have been made by artificial nuclear transmutations. All the actinides are radioactive. Actinides are analogous to lanthanides and involve the filling of 5f-orbitals.
The following general characteristics are shown by actinides:
Electronic configuration: In lanthanides, after lanthanum 4f-orbitals become appreciably lower in energy than the 5d-orbitals. Thus, in lanthanides the electrons fill the 4f-orbitals in a regular way with minor differences where it is possible to attain a half filled shell. Similarly, it might have been expected that after actinium the 5f-orbitals would become lower in energy than the 6d-orbitals. However, for the first four actinide elements, Th, Pa, U and Np the difference in energy between 5f and 6d-orbitals is small. Thus, in these elements (and their ions) electrons may occupy the 5f or the 6d) levels or sometimes both. Later in the actinide series the 5f-orbitals do become appreciably lower in energy. Thus, from Pu onwards the 5f-shell fills in a regular way and the elements become very similar. The most widely accepted electronic configurations of actinides are tabulated below. The general electronic configurations of actinides may be written as:
|
Name of the element |
Symbol |
Atomic number |
Electronic configuration |
Oxidation states |
|
Actinium |
Ac |
89 |
[Rn]6d1, 7s2 |
+3 |
|
Thorium |
Th |
90 |
[Rn]6d2, 7s2 |
+3, +4 |
|
Protactinium |
Pa |
91 |
[Rn]5f2, 6d1, 7s2 |
+3, +4, + 5 |
|
Uranium |
U |
92 |
[Rn]5f3, 6d1, 7s2 |
+3, + 4, + 5, + 6 |
|
Neptunium |
Np |
93 |
[Rn]5f4, 6d1,7s2 |
+3, + 4, + 5, + 6, + 7 |
|
Plutonium |
Pu |
94 |
[Rn]6f6, 6d1, 7s2 |
+3, + 4, + 5, + 6, + 7 |
|
Americium |
Am |
95 |
[Rn]5f7, 6d1, 7s2 |
+3, + 4, + 5, + 6 |
|
Curium |
Cm |
96 |
[Rn]5f7, 7s2 |
+3, + 4 |
|
Berkelium |
Bk |
97 |
[Rn]5f9, 7s2 |
+3, + 4 |
|
Californium |
Cf |
98 |
[Rn]5f10, 7s2 |
+2, + 3 |
|
Einsteinium |
Es |
99 |
[Rn]5f11, 7s2 |
+2, + 3 |
|
Fernium |
Fm |
100 |
[Rn]5f12, 7s2 |
+2, + 3 |
|
Mendelevium |
Md |
101 |
[Rn]5f13, 7s2 |
+2, + 3 |
|
Nobelium |
No |
102 |
[Rn] 5f14, 7s2 |
+2, + 3 |
|
Lawrencium |
Lr |
103 |
[Rn] 5f14, 6d1 7s2 |
+ 3 |
Oxidation states: The known oxidation states of the actinide elements are shown in the above table. The actinides exhibit most common oxidation state of +3 like the lanthanides. However, this state is not always most stable as for the first four elements (Th, Pa, U and Np). For example, U3+ is readily oxidised in air and in solution. +3 state is the most stable state for the later elements Am → Lr (except No). The most stable oxidation states for the first four elements are Th (+4), Pa (+5) and U (+6). The high oxidation states involve using all the outer electrons in including f electrons for bonding. Though Np shows +7 oxidation state but it is oxidising and the most stable state for Np is +5. Pu shows all the oxidation states from +3 to +7 but the most stable is +4. Am shows oxidation states from +2 to +6. Am2+ has an f7 configuration. It is the analogue of Eu2+ but it only exists in solid as fluoride. However, for Am and almost all the remaining elements +3 state is most stable.+4 oxidation state exists for all the elements from Th to Bk.
Cf2+ , Es2+ , Fm2+ Md2+ and No2+ exist as ions in solution. Their properties are like alkaline earth metals particularly Ba2+. It is the most stable state for No and corresponds to an f14 configuration.
+5 oxidation state occurs for the elements Pa → Am. A few solid compounds are known in +5 oxidation state but M5+ ions do not occur in solution. However, MO2+ ions exist between pH 2-4. These ions disproportionate rapidly in solution.
+6 oxidation state occurs as fluorides, MF for the elements U, Np, Pu and Am. The +6 state is more widely found as the dioxoion, MO The ion is stable and exists both in solution and in crystals.
The 5f-orbitals extend into space beyond the & and 6p-orbitals and participate in bonding. This is in direct contrast to the lanthanides where the 4f-orbitals are buried deep inside in the atom, totally shielded by outer orbitals and thus unable to take part in bonding. The participation of the Sf-orbitals explains the higher oxidation states shown by earlier actinide elements.
The lower oxidation states tend to be ionic and the higher ones are covalent. M M and M ions are known. Hydrolysis of these ions occurs quite readily but can be suppressed by using acid solutions. Hydrolysis of compounds in the higher oxidation states gives +5-MO ions and +6 ions.
Physical properties: The elements are all silvery metals. The melting points are moderately high but are considerably lower than those of transition elements. The size of the ions decreases gradually along the series because the extra charge on the nucleus is poorly screened by the f electrons. This results in an ‘actinide contraction’ similar to the lanthanide contraction. Actinides have high densities. Some properties of the actinides up to berkelium are tabulated below. Not much information is available about heavy actinides.
|
Element |
Melting point (*0C) |
Density (g cm–3) |
Radius M3+ (pm) |
Radius M4+ (pm) |
|
Thorium |
1750 |
11.8 |
108 |
94 |
|
Protactinium |
1552 |
15.4 |
104 |
90 |
|
Uranium |
1130 |
19.1 |
102.5 |
89 |
|
Neptunium |
640 |
20.5 |
101 |
87 |
|
Plutonium |
640 |
19.9 |
100 |
86 |
|
Americium |
1170 |
13.7 |
97.5 |
85 |
|
Curium |
1340 |
13.5 |
97 |
85 |
|
Berkelium |
986 |
14.8 |
96 |
83 |
Colour of the ions: Actinide ions are generally coloured. The colour of the ions depends on the number of electrons present in 5f-orbitals. The ions having no electron in 5f-orbitals (i.e.. Sf or seven electrons in 5f-orbitals ( e.,5j are colourless. The ions having 2 to 6 electrons in Sf-orbitals are coloured both in the crystalline and in aqueous solution. The colour is due to f-f transition.
|
Th4+ |
U3+ |
Np3+ |
Pu3+ |
Am3+ |
Cm3+ |
U4+ |
Np4+ |
|
(5f0) |
(5f3) |
(5f4) |
(5f5) |
(5f6) |
(5f7) |
(5f2) |
(5f3) |
|
Colourless |
Red |
Purple |
Violet |
Pink |
Colourless |
Green |
Yellow green |
Magnetic behaviour: Majority of the ions of the actinides possess unpaired electrons, thus they are paramagnetic in nature. Th3+ (5f1), Pa4+(5f1), U3+(5f3), Np5+(5f2), Pu4+(5f4), Am5+(5f4), etc, are paramagnetic. Cations of actinides which contain only paired electrons are diamagnetic, Ac3+(5f0), Th4+ (5f0), U6+ (5f0), Lr3+ (5f14), etc., are diamagnetic in nature.
Formation of complexes: Actinides have somewhat higher tendency to form complex compounds in comparison to lanthanides. This is due to their higher charge and smaller size of their ions. Most of the halides of actinides form complex compounds with alkali metal halides. Actinides form chelates with organic compounds such as EDTA and oxime. The degree of complex formation for the Ions M4+, MO22+, M3+ and MO2+ decreases in the order
Chemical reactivity: On account of low ionisation energies, the actinides are highly electropositive metals. They react with hot water and tarnish in air forming an oxide coating. The metals react readily with HCI but reactions with other acids are slower than expected. Concentrated HNO makes Th, U and Pu passive. The metals react with oxygen, the halogens and hydrogen. Actinides act as strong reducing agents.
Radioactivity: All the actinide elements are radioactive in nature.
Uses of Actinides:
Thorium, uranium and plutonium are three actinides which find uses as such or in the form of compounds.
Uses of thorium: (a) When thorium dioxide containing 1% CeO2 is heated in a gas flame, it emits a brilliant white light. Because of this, it is used for making incandescent gas mantles. The mantle made from silk fibre is treated with a mixed solution of 99% thorium nitrate and 1% cerium nitrate. When this mantle is fixed in the lamp and ignited, the silk fibre burns away leaving behind a network of thoria (ThO2) and ceria (CeO2).
(b) Naturally occurring thorium is almost entirely Th-232. This isotope is not fissionable but is convened into 1.1-233 which is fissionable.
Thus, thorium is used for the production of fissionable material needed for atomic reactors.
(c) ‘Thorium salts are used in medicines for the treatment of cancer.
Uses of uranium: (a) The salts of uranium find use in glass industry (for imparting green colour), textile industry, ceramio industry and in medicines.
(b) The 1.1 isotope is used as nuclear fuel in atomic reactors and atom bombs.
Uses of plutonium: Pu-239 is used as a nuclear fuel. It is obtained from U-238.
FORMULAE AND CONCEPTS AT A GLANCE
1. In d-block elements the differentiating e– enter the d-subshell while in f-block elements they enter f-subshell.
2. d-block elements are called transition elements since all of their properties lie in between s & p block elements. They show a transition in properties from high metallic nature to non-metallic.
3. Fe is most abundant transition metal in earth crust followed by Ti.
4. Only Os and Ru in transition elements show +8 oxidation state.
5. The highest oxidation state of transition metals are formed in their compounds with fluorine and oxygen. This is due to higher electronegativity and small size of F and O atoms.
6. Chlorides of Hg, Ag, Pb and Sn do not give chromyl chloride test.
7. Fe(OH)3 in free state never exists. On hydrolysis, FeCl3 does not give hydroxide but gives Fe2O3(H2O)n.
8. The colour in d-block elements is due to d-d-transitions while in f-block elements, it is due to f-f transitions.
9. Transition metals have very high melting and boiling points due to stronger metallic bonding. The melting points of the transition elements first rise to a maximum and then fall as the atomic number increases.
10. The transition elements readily form alloys with themselves and with other elements (e.g., a copper-tin alloy is used for mirrors, brass is a copper-zinc alloy). Tungsten is used to make tools and filaments in light bulbs.
11. Apart from copper, the transition metals are all white lustrous metals. They vary widely in abundance.
12. Iridium is most dense metal with density ≈ 22.61g cm–3 followed by osmium (≈ 22.57g cm–3).
13. Lower oxidation states of transition metals are chiefly ionic while higher oxidation states are covalent. (Fajan rule).
14. Ti is called as Wonder metal due to its unique and useful properties.
15. Those metals which produce fire when rubbed with any surface or object are known as pyrophoric metals e.g., misch metal.
SOLVED PROBLEMS-1
Prob 1. Why are the ionization energies of 5d elements greater than 3d elements?
Sol: In the 5d series, after lanthanium (Z = 57), there is lanthanide contraction. In each group, the size of 5d element is smaller while nuclear charge is greater than 3d element. Hence, ionization energies of 5d elements are greater than 3d elements.
Prob 2. Give reasons why the lowest oxide of a transition metal (say, chromium, atomic number 24) is basic whereas the highest oxide is usually acidic?
Sol: Lowest oxide of Cr is CrO which is basic. The highest oxide is CrO3 which is acidic (Inbetween, Cr2O3 is amphoteric). Higher the oxidation state of the metal, more easily it can accept electrons and hence greater is the acidic character.
Prob 3. Explain how the colour of K2Cr2O7 solution depends on pH of the solution?
Sol: In the solution, the following equilibrium exists:
Prob 4. In what way do the d-block metals differ from alkali and alkaline earth metals?
Sol:
|
d-block metals |
Alkali and alkaline earth metals |
|
(i) They are less metallic. |
They are more metallic. |
|
(ii) The last electron in them enters d-orbital. |
The last electron in them enters s-orbital. |
|
(iii) Most of them form complexes. |
Very few of them form complexes. |
|
(iv) Their salts are mostly coloured. |
Their salts are white. |
|
(v) They are usually used as catalyst. |
Very few of them are used as catalysts. |
Prob 5. Write equation to show how H2O2 reduces to Mn2+ in acidic solution.
Sol:
Prob 6. Silver chloride dissolves in excess ammonia, why?
Sol: AgCl forms a soluble complex with
Prob 7. Why hydrated copper sulphate is blue while anhydrous copper sulphate is white?
Sol: In hydrated copper sulphate, four water molecules are present as ligands. In the presence of these ligands d-orbitals split into two sets of slightly different energies. Hence d-d transition takes place absorbing red wavelength. In anhydrous CuSO4, d-orbitals remain degenerate. Hence, no d-d transition can occur. The white light is completely reflected back. Hence, it looks white.
Prob 8. Explain why mercury (I) ion exsts as ion while copper (I) ion exists as Cu+ ion.
Sol: The electronic configuration of Hg(I), i.e., Hg+ is [Xe]4f145d10 6s1 and thus has one electron in the valence 6s-orbital. If this were so, all Hg (I) compounds should be paramagnetic but actually they are diamagnetic. This behaviour can be explained if we assume that the singly filled 6s-orbitals of the two Hg+ ions overlap to form a Hg-Hg covalent bond. Thus, Hg+ ions exist as dimeric species, i.e., . In contrast, the electrons to form dimeric species, i.e., and hence it always exists as Cu+ ion.
Prob 9. HgCl2 and SnCl2 cannot exists together in an aqueous solution. Why?
Sol: SnCl2 is a strong reducing agent and hence reduces HgCl2 first to Hg2Cl2 (white) and then to Hg (black)
Prob 10. In moist air copper corrodes to produce a green layer on the surface. Explain.
Sol: In presence of moist air, a thin film of green basic copper carbonate is formed on its surface and hence copper corrodes.
Basic copper carbonate (green)
SOLVED PROBLEMS-2
Prob 1. The most stable oxidation state of + 3 is shown by
(A) Mn (B) Co (C) Ni (D) Fe
Sol: (D) Fe3+ [Ar]3d5. Half filled d-subshell is very much stable.
Prob 2. The dichromate ion is in equilibrium with chromate ion in aqueous solution as
The oxoanion has
(A) same oxidizing property in acidic and alkaline solution
(B) better oxidizing property in acidic solution
(C) better oxidizing property in alakaline solution
(D) no oxidizing property in acidic or alkaline solution
Sol: (B) In acidic
Prob 3. When a salt X is added to the alkaline solution of red vapour obtained in the above question, brick precipitate is formed. The precipitate is
(A) BaCrO4 (B) PbCrO4 (C) Ag2CrO4 (D) CaCrO4
Sol: (D) CrO2Cl2+4NaOH Na2CrO4 + 2NaCl + 2H2O
Na2CrO4 + 2AgNO3 2NaNO3 + Ag2CrO4 (brick red ppt)
Prob 4. The magnetic moment of a paramagnetic species is given by (n= number of unpaired electrons, S = total spin)
(A) (B) (C) (D)
Sol: (D) Magnetic moment =
Prob 5. The total spin and paramagnetism (BM) of ferrocyanide ion are respectively
(A) (B) (C) (D)
Sol: (D) O.N. of Fe in ion = + 2;
Configuration of Fe(II) = 3d6; n = 4
Prob 6. Among the following compounds, which is thermally stable?
(A) FeCO3 (B) Fe3O4 (C) FeO (D) Fe2O3
Sol: (D)
Prob 7. The correct statement among the following is
(A) FeI3 is stable is aqueous solution.
(B) An acidified solution of K2CrO4 gives yellow precipitate on mixing with lead acetate.
(C) The species exists but does not.
(D) Both copper (I) and copper (II) salts are known in aqueous solution.
Sol: (D) I– ion is a stronger reducing agent than CI– ion. It reduces Cu2+ ion to Cu+ ion.
Thus, CuI2 is reduced to CuI and the species (CuI4)2– does not exist.
Prob 8. On heating AgNO3 above its melting point, the gas evolved is
(A) NO2 only (B) NO2 and O2 (C) O2 only (D) N2 and O2
Sol: (D) (above the melting point)
Prob 9. The compound that gets oxidized even on exposure to air, is
(A) FeSO4. (NH4)2SO4 (B) Hg2Cl2 (C) FeSO4.7H2O (D) Cu2Cl2
Sol: (D) Hydrated ferrous sulphate acquires brownish-yellow colour due to the formation of basic ferric sulphate by atmospheric oxygen.
Prob 10. When SnCl2 and HgCl2 in the mole ratio of 1 : 2 are mixed in aqueous solution, then which of the following is obtained?
(A) Hg (B) Hg2Cl2 (C) FeSO4.7H2O (D) Cu2Cl2
Sol: (D)