








Introduction
As the name implies, hydrocarbons are organic compounds containing carbon and hydrogen only. As fuels, hydrocarbons play a key role in our daily life. The following are the familiar examples of fuels: LPG (liquefied petroleum gas), CNG (compressed natural gas) and LNG (liquefied natural gas). This is also a fuel, obtained by liquefaction of natural gas.
Petrol, diesel, and kerosene oil are obtained by the fractional distillation of petroleum found under the earth’s crust. Coal gas is obtained by the destructive distillation of coal. Natural gas is found in upper strata during drilling of oil wells. The gas after compression is known as CNG. LPG is used as a domestic fuel with the least pollution. Kerosene oil is also used as a domestic fuel but it causes some pollution. Automobiles need fuels like petrol, diesel and CNG. Petrol and CNG operated automobiles cause less pollution.
All these fuels are basically a mixture of hydrocarbons. Thus hydrocarbons are the important sources of energy. Hydrocarbons are also used for the manufacture of polymers like polythene, polypropene, polystyrene, etc. Higher hydrocarbons are used as solvents for paints. Hydrocarbons are also used as the starting materials for manufacture of many dyes and drugs.
Classification
On the basis of carbon skeleton, hydrocarbons are divided into two main classes:
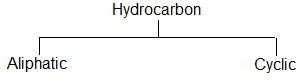
Aliphatic hydrocarbons are open-chain or acyclic compounds. Depending upon the types of carbon – carbon bonds present, they can be further classified into two main categories:
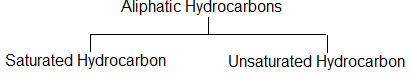
Saturated aliphatic hydrocarbons contain carbon-carbon and carbon-hydrogen single bonds, while unsaturated aliphatic hydrocarbons contain carbon-carbon multiple bonds – double bonds, triple bonds or even both.
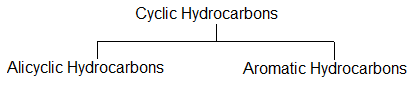
Cyclic hydrocarbons are divided into two main classes
ALKANES
Saturated aliphatic (or acyclic) hydrocarbons are called alkanes. Many occur naturally. The chief source of the alkanes is mineral oil or petroleum, which occurs in many parts of the world.
The simplest member of the alkane family and, indeed, one of the simplest of all organic compounds is methane, CH4. It is an end product of the anaerobic (“without air”) decay of plants (i.e., of the breakdown of certain very complicated molecules). It is also the major constituent (up to 97%) of natural gas. It is the dangerous fire damp of the coal mine, and can be seen as marsh gas bubbling to the surface of swamps.
These hydrocarbons are chemically inert under normal conditions as they do not react with commonly available reagents. Hence, they were earlier known as paraffins (Latin: parum, little; affinis, affinity). The general formula for alkanes is CnH2n + 2, where ‘n’ is the number of carbon atoms.
Shape of alkanes
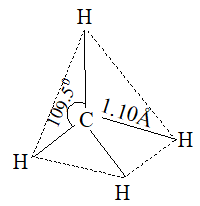
In methane each of the four hydrogen atoms is bonded to the carbon atom by a normal covalent bond. Whenever carbon is bonded (covalently) to four other atoms, its bonding orbitals are sp3 hybrid orbitals and are directed towards the corners of a regular tetrahedron.
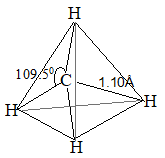
This way the orbitals are as far apart as possible [VSEPR theory]. Thus, methane has a tetrahedral shape, verified by electron diffraction study.
In higher alkanes, two or more tetrahedra are joined together in which C – C and C – H bond lengths are 153 – 154pm and 110 – 112pm.
C – C and C – H s bonds are formed by head-on overlapping of sp3 hybrid orbitals of carbon and 1s orbitals of hydrogen atoms.
Nomenclature of alkane
We have already discussed about nomenclature of different classes of organic compounds in Phase II.
Example What is the IUPAC name of the following alkane?
Strategy Find the longest continuous carbon chain in the molecule……..?
Isomerism in alkanes
All the alkanes can be derived formally from methane by substituting hydrogen atoms by methyl groups (CH3) :
The next molecules will be C4H10 C5H12, etc. when the alkane contains three or more carbon atoms substitution gives rise to isomerism. For example, propane, C3H8, can give rise to two butanes, C4H10 : n – butane (butane) by substitution at a terminal carbon atom or primary carbon atom, and isobutane (2 – methyl propane) by substitution at the central carbon atom or secondary carbon atom.
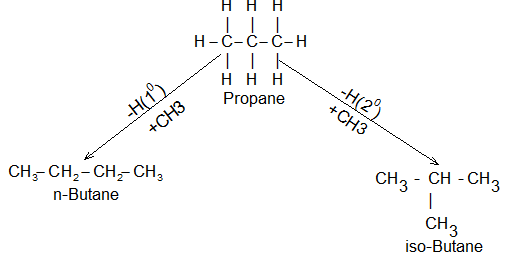
Similarly substitution of hydrogen atoms of butanes by methyl groups gives rise to three pentanes:
| Pentane (n-pentane) | 2 – Methylbutane (isopentane) | 2,2 – Dimethylpropane (neopentane) |
As the number of carbon atoms in the alkane increases, the number of possible isomers increases rapidly, e.g., the alkane C15H32 can exist in 4,347 isomeric forms!
Illustration1: Write structures of different isomeric alkyl groups corresponding to the molecular formula C5H11. Also give IUPAC names of alcohols obtained by attachment of –OH groups to these alkyl groups.
Solution: Remove all possible different H atoms from three isomeric pentanes, and attach -OH groups to the carbons.
Alkyl groups derived from n-pentane and corresponding alcohols:
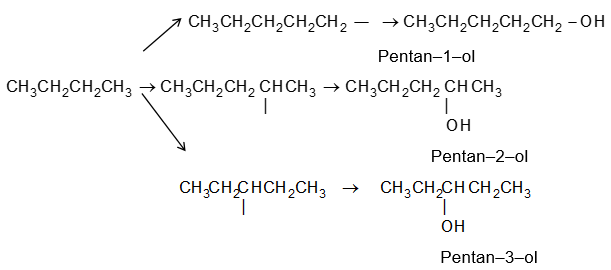
Alkyl groups derived from isopentane and corresponding alcohols:
Alkyl group derived from neopentane and corresponding alcohol:
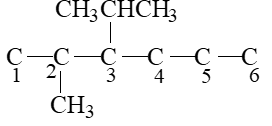
2, 2-Dimethylpropan-1-ol If it is important to write the correct IUPAC name for a given structure, it is equally important to write the correct structure from the given IUPAC name. Following steps should be noted as illustrated for 3-ethyl-2, 2–dimethylpentane.
Step 1 Write the longest continuous chain of carbon atoms corresponding to the parent alkane:
Step 2 Number the parent chain
Step 3 Attach the substituents to the correct carbon atoms
Step 4 Satisfy the valence of each carbon atom by putting the correct number of hydrogen atoms.
Illustration 2: Draw the structure of 3–isopropyl–2–methylhexane. This is the reverse of previous example and employs a reverse strategy. First draw the carbon skeleton corresponding to the parent name (hexane) and the identify then substituents and attach them.
Solution: Draw the skeleton of the parent compound (hexane):
Hexane
Now place the substituents (3- isopropyl and 2-methyl) on the proper carbons:
Finally add hydrogen to complex the structure:
Illustration3: Write the correct IUPAC names of the following:
a) 3 – Ethylpentane
b) 5 – Ethyl – 3- methylheptane
Solution:
a)
Notice that the longest continuous carbon chain is of six carbon atoms (not that of five). Thus, correct name is 3 – methylhexane.
b)
Ethyl group takes precedence over methyl group alphabetically. Thus, numbering is to be started from the end which gives lower number to ethyl group. Hence, correct name is 3–ethyl – 5 – methylheptane.
General methods of preparation of the alkanes:
Each of the smaller alkanes, from methane through n–pentane and isopentane, can be obtained in pure form by fractional distillation of petroleum and natural gas; neopentane does not occur naturally. Above the pentanes, the number of isomers of each homolog becomes so large and the boiling point differences become so small that it is no longer feasible to isolate individual, pure compounds, these alkanes must be synthesized by one of the methods discussed below:
1) From unsaturated aliphatic hydrocarbons:
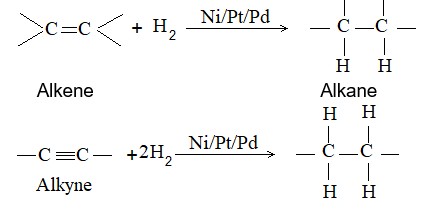
Examples:
Propene Propane
But-2-yne n–Butane
Dihydrogen gas adds to alkenes and alkynes in the presence of finely divided catalysts like Raney nickel, platinum or palladium to form the corresponding alkanes. This process is called catalytic hydrogenation. These metals adsorb the dihydrogen gas and the unsaturated hydrocarbon on their surface and activate the reduction.
Formerly, many organic compounds were reduced by passing their vapours mixed with dihydrogen gas over nickel (on a suitable inert, porous support) heated at 200 – 3000C. Any reduction that is carried out in this manner is referred to as the Sabatier–Senderens reduction, in honour of the workers who first introduced this method.
Raney nickel (prepared by the method introduced by Raney, 1927) is more reactive than the supported nickel catalyst, and is usually effective at lower temperatures, often at room temperature. Alkenes may also be reduced to alkanes by a variety of chemical reagents.
A very useful method for preparing long-chain alkanes involves conversion of suitable alkenes into alkylboranes followed by coupling of alkylboranes by means of silver nitrate in the presence of sodium hydroxide at room temperature:
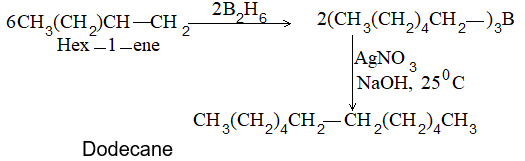
Unlike boranes may also be coupled.
If the alkylborone is treated with a carboxylic acid such as propionic acid, the corresponding alkane is obtained by protolysis:
2. From alkyl halides:
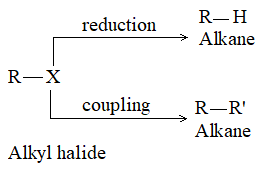
a) Reduction of alkyl halides
It simply involves the replacement of a halogen atom by a hydrogen atom, the carbon skeleton remains unchanged. It can be done directly or indirectly via the Grignard reagent. Direct reduction of alkyl halides may be done in different ways:
i) Reduction by metals dissolved in suitable solvents
- Zinc and hydrochloric acid (dilute)
- Zinc and acetic acid
- Zinc and sodium hydroxide
- Zinc – copper couple and ethanol
Mechanism There is an electron – transfer from the metal to the substrate, followed by the addition of proton from the solvent:
(from solvent)
Examples:
ii) Reduction by hydride transfer reagents
| Alkyl halide/Reagent | Lithium aluminium hydride (LiAlH4) | Sodium borohydride (NaBH4) | Triphenyl tin hydride Ph3SnH |
| RCH2X, primary alkyl halide | ✓ | ✗ | ✓ |
| R2CHX, secondary alkyl halide | ✓ | ✓ | ✓ |
| R3CX, tertian alkyl halide | ✗ | ✓ | ✓ |
iii) Catalytic hydrogenolysis It involves the addition of dihydrogen gas to sigma bonds in the presence of a suitable catalyst.
Primary, secondary and tertiary alkyl halides (iodides, bromides and chlorides) are converted into the corresponding alkanes by catalytic hydrogenolysis. The best available catalyst is Pd–C, but Raney Ni is also effective provided it is used in large amounts.
Examples
iv) Reduction with concentrated hydriodic acid Iodides may be reduced into the corresponding alkanes by heating with concentrated hydriodic acid at 1500
Examples:
Reduction with conc. HI is usually carried out in the presence of a small amount of red phosphorus which regenerates the hydriodic acid from the iodine formed.
The hydriodic acid – red phosphorus mixture is one of the most powerful reducing agents used in organic chemistry.
v) Indirect reduction of alkyl halides is done via the Grignard reagents.
A solution of an alkyl halide in dry ethyl ether, (C2H5)2O, reacts vigorously with the turnings of metallic magnesium to form a solution of Grignard reagent (after Victor Grignard of the University of Lyons)
Examples:
The Grignard reagent is one of the most useful and versatile reagent known to the organic chemists. It has the general formula RMgX (or ArMgX), and the general name alkyl magnesium halide (or aryl magnesium halide). It is a typical ionic compound.
The alkyl group remains unchanged during the preparation of the reagent. Thus n-propyl iodide gives n-propylmagnesium iodide:
The Grignard reagent is the best–possible example of organometallic compounds. Generally speaking, organometallic compounds are those organic compounds in which a metal (lithium, potassium, sodium, zinc, mercury, lead, thallium etc.) is directly bonded to carbon. Since almost any metal is less electronegative than carbon, the carbon metal bond in organometallic compounds is highly polar. Thus, the organic group has considerable carbanion character. The amount of polar character of the carbon metal bond depends on the nature of the metal:
(the order of polarity)
Sodium and potassium alkyls are salts.
Grignard reagents, on treatment with compounds more acidic than an alkane, are decomposed to alkanes:
Examples
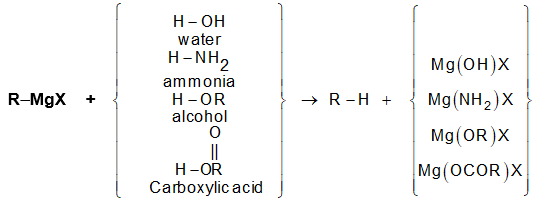
For the preparation of an alkane, one acid is as good as another, so we naturally select water as the most available and convenient.
b) Coupling of alkyl halides
It can be done either through the Wurtz reaction or by the Corey-House synthesis.
i) Wurtz reaction:
It involves coupling of alkyl groups when a solution of an alkyl halide (preferably the bromide or idodide) in dry ether (free from moisture and alcohol) is treated with sodium metal:
where R and R’ are the two unspecified alkyl groups which may or may not be the same. The equation given above clearly shows that in addition to the desired alkane R – R’, there will also be formed, the alkanes R – R, and R’ – R’. Besides, unsaturated hydrocarbons are also formed.
Limitation The best yield of an alkane will be obtained when R and R’ are the same, i.e., when the alkane contains an even number of carbon atoms and is symmetrical. Experimentally, it is found that the Wurtz reaction gives good yields only for even carbon alkanes of high molecular mass. It fails with tertiary alkyl halides.
Application: Wurtz reaction is used to ascend the homologous series through the preparation of higher alkanes containing even number of carbon atoms.
Examples
Though sodium is extensively used in the Wurtz reaction, other metals such as Ag, Cu may also be used in a finely divided state.
Mechanism: Two possibilities have been suggested for the Wurtz reaction
1) (Intermediate) formation of an organo-metallic compound:
Step 1 :
Step 2 :
Step 2 is typically a bimolecular nucleophilic n-butane displacement.
Side products, ethane and ethene, may be formed as follows (dehydrohalogenation):
It is typically a bimolecular elimination mechanism.
2) The free – radical mechanism has been found to be operational in some cases
Disproportionation [i.e., intermolecular hydrogenation, one particle acquiring hydrogen at the expense of the other] of free radicals accounts for the formation of ethane and ethylene as the side products:
ii) Corey – House synthesis
It involves coupling of alkyl halides with organometallic compounds. For this purpose, an alkyl lithium, RLi, is prepared from an alkyl halide, RX, in much the same way as a Grigard reagent:
Alkyl lithium
To it is added cuprous halide, CuX, leading to the formation of a lithium dialkylcopper, R2CuLi:
Lithium dialkylcopper
Finally is added the second alkyl halide, R’X. Coupling takes place in the reaction between a lithium dialkylcopper, R2CuLi, and an alkyl halide, R’X (R’ stands for an alkyl group that may be same as, or different from R):
Alkane
Ultimately the alkane is synthesized from the two alkyl halides, RX and R’X.
For good yields, the alkyl group R in the organometallic may be primary, secondary, or tertiary but R’X should be a primary alkyl halide:
Examples:
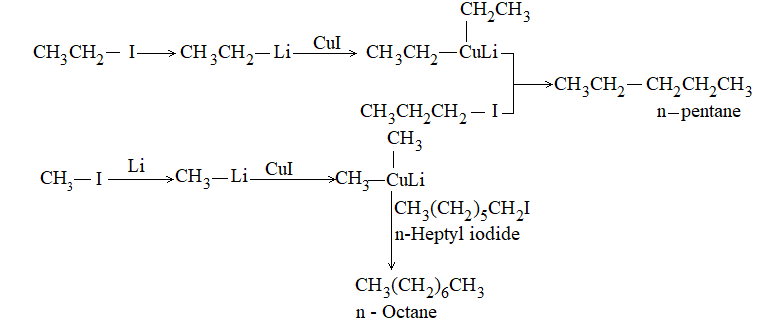
It is clear that the alkyl group R is transferred from copper, taking a pair of electrons from it, and becomes attached to the alkyl group R¢ in place of halide ion (nucleophilic aliphatic displacement).
3) From saturated monocarboxylic acids
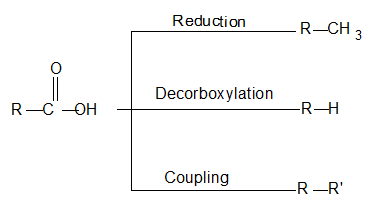
i) Reduction of carboxylic acids
Heating saturated monocarboxylic acids with hydrogen iodide – red phosphorus under pressure, or with dihydrogen gas under pressure at elevated temperature in the presence of a nickel catalyst, produces an alkane:
Examples
ii) Decarboxylation
When the anhydrous sodium salt of a saturated monocarboxylic acid is heated with sodalime [a mixture of sodium hydroxide and calcium oxide], an alkane containing one carbon atom less than the acid and other products are formed:
Examples:
This process of eliminating carbon dioxide from the carboxyl group of a carboxylic acid is known as decarboxylation.
Limitations Only sodium acetate decomposes according to the equation given above. In rest of the cases tested ¾ propionate, butyrate and caproate various products are obtained. For example:
This is therefore not a useful general method for the preparation of alkanes, since the separation of the products is usually difficult.
Mechanism
Salts decompose by an reaction.
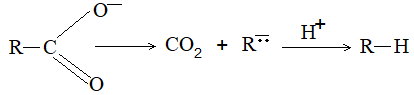
Thermal decomposition of free acids:
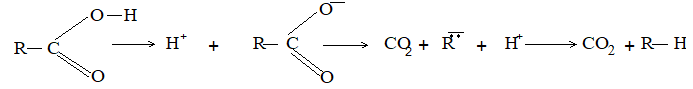
iii) Coupling through Kolbe’s electrolytic method:
It involves the electrolysis of concentrated aqueous solution of sodium or potassium salt of a saturated monocarboxylic acid or mixture of saturated monocarboxylic acids:
[At cathode + R-R’+2CO2] At anode
Example:
If R and R’ are different, then hydrocarbons R–R and R’-R’ are also obtained, such mixtures can often be separated readily. The by-products are alkenes, alcohols (in alkaline medium) and esters.
The Kolbe electrolytic method has application in the synthesis of natural compounds, particularly lipids.
Mechanism: The reaction follows the free – radical path:
At anode: The carboxylate ion discharges at the anode to form a free radical.
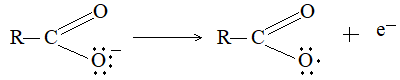
The carboxylate free radical then breaks up into the alkyl free radical and carbon dioxide:
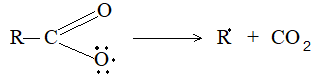
Finally two alkyl free radicals couple together to form a higher alkane:
At cathode:
Water has higher reduction potential than sodium, thus, water undergoes reduction to form the dihydrogen gas:
The presence of Na+(aq) and OH–(aq) leads to the formation of NaOH (aq).
Limitation: Methane can’t be prepared by this method.
Extension: Kolbe’s electrolytic method can be extended to make alkenes as well as alkynes:
Conc. aqueous solution of sodium or potassium salt of Alkene a saturated dicarboxylic acid
Example:
Concentrated aqueous solution of sodium or Alkyne potassium salt of an unsaturated dicarboxylic acid
Example:
4) From carbonyl compounds
Aldehydes and ketones may be reduced to corresponding alkanes:
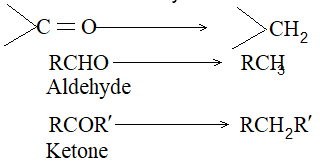
The following methods are employed:
i) By heating with concentrated hydriodic acid in the presence of a small amount of red phosphorus.
ii) With amalgamated zinc and concentrated hydrochloric acid (Clemmensen reduction):
H+ comes from hydrochloric acid and e– is provided by zinc.
The Clemmensen reduction does not work well for aldehydes but is reasonably good for many ketones.
iii) Heating hydrazones (or semicarbazones) of aldehydes and ketones with sodium ethoxide at 1800C [Wolff-Kishner reduction]:
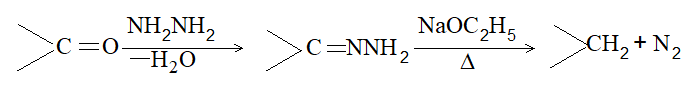
Example:
The yield of hydrocarbon is better than that obtained by the Clemmensen reduction.
5) From monohydric alcohols
Primary, secondary and tertiary monohydric alcohols may be reduced into the corresponding alkanes by heating with concentrated hydriodic acid in the presence of a small amount of red phosphorus:
Examples:
Exercise 1:
(i) The compound which has one isopropyl group is
(A) 2, 2, 3, 3-tetramethylpentane
(B) 2, 3-dimethylpentane
(C) 2, 2, 3-trimethylpentane
(D) 2-methylpentane
(ii) Formation of alkane by action of zinc and alkyl halide is called
(A) Wurtz reaction
(B) Frankland reaction
(C) Kolbe’s reaction
(D) Clemmensen reaction
(iii) When water vapours are passed over aluminium carbide, we get
(A) acetaldehyde
(B) ethylene
(C) methane
(D) methyl alcohol
Physical properties of the alkanes
Physical states: Due to the weak van der Waals forces, the first four normal alkanes (C1 to C4) are colourless and odourless gases, the next thirteen normal alkanes (C5 to C17) are colourless and odourless liquids, and from C18 onwards, colourless and odourless solids at 298K and 1 atm pressure.
Boiling points: The boiling points of the normal (unbranched) alkanes show a regular (steady) increase with increasing molecular mass. Except for the very small alkanes (C1 to C4), the boiling point rises 20 to 30 degrees for each carbon that is added to the chain.
Melting points: The normal alkanes do not exhibit the same smooth increase in melting points with increasing molecular mass that they show in their boiling points. There is an alternation as one moves from a normal alkane with an even number of carbon atoms to the next one with an odd number of carbon atoms. For example, n –propane (mp – 1870C) melts lower than ethane (mp – 1720C) and also lower than methane (mp – 1830C). n – Butane (mp – 1380C) melts 490C higher than propane but only 80C lower than pentane (mp – 1300C). Hence there is a saw-tooth pattern. If, however, the even-and odd-numbered alkanes are plotted on separate curves, there is a smooth increase in melting point with increasing molecular mass.
Effect of branching on boiling and melting points: In a group of isomeric alkanes, the normal compound always has the highest b.p. and m.p. Generally, the greater the branching, the lower is the b.p. Thus n-butane has a boiling point of 00C and isobutane -120C. n-Pentane has a boiling point of 360C, isopentane with a single branch 280C, and neopentane with two branches 9.50C. Similarly n-hexane boils at 68.70C, and 2-methyl pentane and 3-methylpentane, each having one branch, boil lower at 60.3 and 63.30C, respectively. 2, 3– Dimethylbutane and 2, 2-dimethylbutane each with two branches boil lower, at 58 and 49.70C, respectively. The effect of chain branching on the melting points of alkanes is not easy to predict. However, the chain-branching producing highly symmetrical structures results in abnormally high melting points. For example, the compound 2, 2, 3, 3 – tetramethylbutane (mp 100.70C) melts 43.70C higher than n-octane (mp – 570C) but boils 200C lower than it.
Solubility: Alkanes are almost completely insoluble in water. Liquid alkanes are soluble in one another and they generally dissolve in non-polar solvents and solvents of low polarity. Examples of good solvents for alkanes are benzene, carbon tetrachloride, chloroform and ether. The solubility diminishes with increase in molecular mass.
Density: As a class, the alkanes are the least dense of all groups of organic compounds. The density also increases with molecular mass of the alkanes, but tends to level off at about 0.79 g mL–1. Thus all alkanes are considerably less dense than water (1.00 g mL–1, the density of water at 40C). As a result, petroleum (a mixture of hydrocarbons) floats on water.
General chemical properties of the alkanes
The alkanes are sometimes referred to by the old- fashioned name of paraffins. This name arose through contracting the two Latin words ‘parum affins’ which implies little affinity or not enough affinity. This name was suggested because alkanes were apparently very unreactive. But reactivity depends upon the choice reagent. Under ‘ordinary’ conditions, the alkanes are inert toward reagents such as acids, alkalis, oxidizing reagents, reducing reagents, etc., but are reactive if the right conditions are used.
Alkanes are inert toward hydrochloric and sulfuric acids, they react readily with acids like HF-SbF5 and FSO3H-SbF5 (“magic acid”) to yield a variety of products. Alkanes are inert toward oxidizing agents like potassium permanganate or sodium dichromate, they get oxidized by halogens. Certain yeasts feed happily on alkanes to produce proteins.
Alkanes mainly undergo substitution reactions in which one or more hydrogen atoms of alkanes are replaced by suitable atoms or groups of atoms. These substitution reactions proceed through free-radical chain reactions, which take place under vigorous conditions and usually yield mixtures of products.
1) Substitution reactions
a) Halogenation It involves substitution of hydrogen of an alkane by a halogen. It takes place either at higher temperature (250 – 4000C, thermal halogenation) or in the presence of diffused sunlight (photohalogenation)
Relative Order of reactivity
H atom of alkane:
Examples:
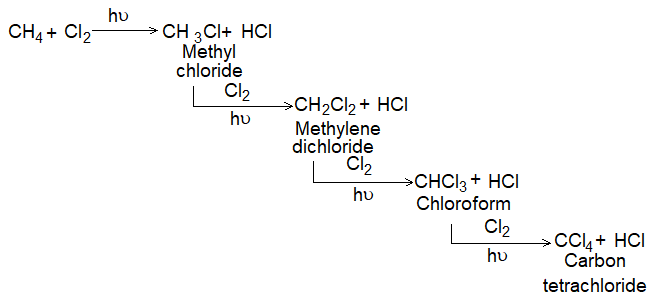
If we restrict the reaction to monohalogenation then,
The extent of halogenation depends largely on the amount of halogen used. A mixture of all possible isomeric mono and polyhalides is obtained, but the isomers are not formed in equal amounts, due to the difference of the reactivity of hydrogen atoms of alkanes.
Reaction with fluorine is so violent that it needs to be controlled by diluting it with an inert gas, and by using an apparatus designed to carry away the heat produced. Chlorination and bromination are not so vigorous. On the other hand, reaction with iodine is so slow that practically there is no reaction. Moreover iodination is reversible:
Thus, it needs to be carried out in the presence of an oxidizing agent, such as HNO3, HIO3 or HgO, which oxidizes the hydrogen iodide (reducing agent) as soon as it is formed and thus drives the reaction to the right:
Fluorides are more conveniently prepared by heating organic chlorides (or bromides) with inorganic fluorides (Swarts reaction) such as AgF, Hg2F2, SbF3 or AsF3:
Iodides are more conveniently prepared by treating the organic chlorides (or bromides) with sodium iodide in acetone (or methanol) solution (Finkelstein or Conant–Finkelstein reaction):
Example:
Mechanism The halogenation of alkanes is an example of a chain reaction, a reaction that involves a sequence of steps, each of which creates a reactive intermediate that brings about the next step. While different chain reactions may vary widely in their details, they all have certain fundamental features in common:
Chain–initiating step It involves homolysis of X-X bond
1.
In this step energy is absorbed and a reactive particle called free radical is created.
Chain propagating steps There are more than one chain-propagating steps, each of which consumes a reactive particle and generates another; here they involve the reaction of halogen atom with an alkane, say methane (step 2) and of alkyl free radical, say methyl radical, with halogen (step 3).
2)
3)
Then (2), (3), (2), (3), etc., until finally a chain is terminated.
The propagation steps (2) and (3) are those which directly give principal products but many other propagation steps are possible. Two such steps given below explain how more highly halogenated products are formed:
Chain terminating steps
Finally through these steps reactive particles are consumed but not generated. The termination of the chain reaction may take place by adsorption of the halogen atoms on the walls of the containing vessel, or by two halogen atoms combining with each other to form a halogen molecule. Another termination is the combination of with or with :
4) or
5) or
6)
Under given set of conditions, about 10,000 molecules of methyl halide are formed for every quantum of light absorbed. Each quantum cleaves one halogen molecule to form two halogen atoms, each of which starts a chain. On the average, each chain consists of 5000 repetitions of the chain-propagating cycle before it is finally stopped.
b) Nitration:
Under suitable conditions, alkanes react with nitric acid resulting into substitution of one or more hydrogen atoms by a nitro-group, NO2. This process which is called direct nitration or just nitration is used to prepare nitro-compounds on a large scale [Industrial preparation]. It is carried out in two ways:
i) Liquid–phase nitration: Here the alkane is heated with concentrated nitric acid under pressure at 1400 It is always a slow process and results in the formation of a large amount of poly-nitro compounds.
ii) Vapour – phase nitration: Nitration of the alkanes may be carried in the vapor phase by heating with nitric acid (or with nitrogen oxides or with dinitrogen) at 150-4750 Each alkane has its optimum temperature for nitration. During vapour-phase nitration, a mixture of mononitroalkanes is obtained. The mixture consists of all the possible mononitroderivatives including those which are including those which are formed by every possible chain fission of the alkane. For example, vapour-phase nitration of n-propane gives a mixture of four mononitroalkanes:
Like halogenation, the different hydrogen atoms in propane are not substituted with equal ease. The ease of substitution by the nitro group is:
Also, any alkyl group present in the alkane can be replaced by a nitro group, through i.e., chain fission.
Vapour-phase nitration is more satisfactory than liquid–phase itration.
Limitation: A mixture of mononitroalkanes is obtained.
Mechanism: Vapour-phase nitration proceeds by a free-radical mechanism.
The better method to prepare nitroalkanes is to heat an alkyl halide with silver nitrite in aqueous ethanolic solution:
c) Sulphonation: The process of sulphonation of an alkane involves substitution of a hydrogen atom by a sulphonic acid group, SO3H, leading to the formation of sulphonic acids. The aliphatic sulphonic acids are named either as alkylsulphonic acids or as alkanesulphonic acids:
When we name the compound as alkanesulphonic acid, the sulphonic acid group is considered as a substituent.
Lower alkanes do not undergo direct sulphonation. Sulphonation of an unbranched alkane from hexane onwards may be carried out by heating the alkane with fuming sulphuric acid (oleum)
The ease of substitution of hydrogen atoms is
30H > 20H > 10H
Substitution of a tertiary hydrogen is very much greater than secondary hydrogen, that of secondary greater than primary and of primary hydrogen atom is very slow.
In n-hexane, the sulphonic acid group enters the chain, mainly at the second carbon atom:
Isobutane containing a tertiary hydrogen atom is readily sulphonated to give t-butylsulphonic acid:
Mechanism: Like halogenation and nitration, it also follows free-radical mechanism.
Sulphuryl chloride (or a mixture of chlorine and sulphur dioxide) in the presence of light or peroxide at 40 – 600, converts alkanes into sulphonyl chloride, often in high yield.
The sulphonyl chloride group enters mainly at the second carbon atom of a normal alkane.
A better method to prepare sulphonic acids is to heat an alkyl halide with sodium sulphite (Strecker reaction)
2) Oxidation
There are two ways to oxidize alkanes:
a) Complete (or uncontrolled ) oxidation
All alkanes readily burn on heating in excess of air or oxygen and get completely oxidized to carbon dioxide and water with the evolution of large amount of heat. This process is called combustion of alkanes.
The general combustion equation for any alkane is:
Due to the release of large amount of heat during combustion, alkanes are used as fuels. For example, the combustion of alkanes is the chief reaction occurring in the internal combustion engine. The mechanism of combustion proceeds through a free radical chain reaction. The reaction is extremely exothermic but requires a very high temperature (say of a flame) for its initiation. During incomplete combustion of alkanes with limited amount of air or oxygen, carbon black is formed which is used in the manufacture of ink, printer ink, black pigments and as filters:
b) Controlled oxidation:
On heating with a controlled amount of air or dioxygen at high pressure or temperature and in the presence of suitable catalysts alkanes undergo controlled oxidation to give different useful products.
Oxidising reagents such as potassium permanganate normally do not oxidize alkanes but they readily oxidize a tertiary hydrogen atom to a hydroxyl group. For example:
3) Isomerization:
It involves conversion of n-alkanes into branched-chain alkanes carrying a methyl group as a side-chain. It is carried out by heating the unbranched alkane with anhydrous aluminium chloride at 3000C. For isomerisation to be feasible a trace of water (to form HCl from AlCl3) together with a trace of alkyl halide or an alkene (to form a carbocation) is essential.
Mechanism: The isomerisation proceeds through an ionic chain reaction:
Step 1: Conversion of alkene ‘impurity’ into a carbonium ion (by the AlCl3 and HCl). This initiates the chain reaction:
Step 2: Isomerisation by a 1, 2 – shift involving a hydride ion:
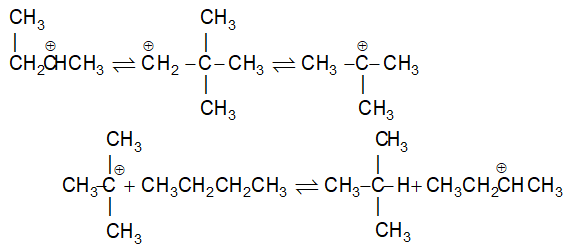
4) Pyrolysis (cracking):
The thermal decomposition (i.e., decomposition by the action of heat alone) of an organic compound is known as pyrolysis. This word originates from the Greek pyr (fire), and lysis (a loosing) and hence means “cleavage by heat”.
Hydrolysis – cleavage by water:
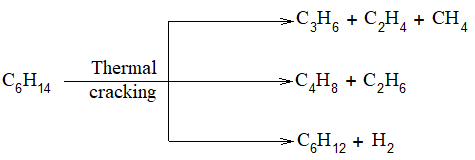
The pyrolysis of alkanes, particularly petroleum, is known as cracking. In thermal cracking alkanes are simply passed through a chamber heated to a high temperature. When heated to about 400 – 6000C, higher alkanes are decomposed into smaller alkanes, alkenes and some dihydrogen (H2) gas:
Higher alkane Smaller alkanes + Smaller alkenes + H2
The actual products obtained from a given alkane depend on:
a) the structure of the alkane,
b) the pressure under which cracking is carried out, and
c) the presence or absence of catalysts such as silica-alumina, silica-alumina-thoria, silica-alumina-
Example:
Mechanism: Cracking of alkanes is a free-radical reaction as many alkanes produce alkyl free radicals of cracking temperatures:
Preparation of oil gas or petrol gas from kerosene oil or petrol involves the principle of pyrolysis. For example, dodecane, a constituent of kerosene oil on heating to 973K in the presence of Pt, Pd or Ni gives a mixture of heptane and pentene:
5) Hydroforming (or catalytic reforming or aromatization):
It involves conversion of normal alkanes having six or more carbon atoms into benzene and its derivatives. It is carried out under pressure at 480-5500C in the presence of oxides of vanadium, molybdenum or chromium on an alumina support. The process is based on the following sequence:
Dehydrogenation Cyclisation Aromatization Isomerisation
The aromatic compounds obtained contain the same number of carbon atoms as the aliphatic starting materials as shown by the following examples:
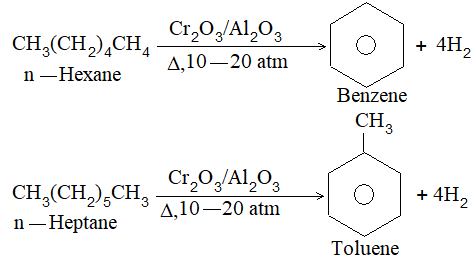
Illustration 4: What are the aromatic compounds formed when n-octane is subjected to hydroforming?
Solution:
6) Steam reforming:
Here the alkane is mixed with steam and a limited supply of oxygen (not air, to avoid the entry of nitrogen). The mixture is passed over a nickel catalyst at a temperature of about 9000C and under a pressure of 30 atmospheres. This converts the alkane into a mixture of carbon monoxide and dihydrogen gas according to the following reaction:
Dihydrogen gas is made in large amounts by the steam reforming of methane. This is the principal method in countries possessing an abundant supply of natural gas.
Methane:
It is the simplest member of the alkane family and also one of the simplest of all organic compounds.
1. Shape: In methane each of the four hydrogen atoms is bonded to the carbon atom by the sharing of a pair of electrons (i.e., by a covalent bond). In every alkane, carbon exhibits sp3 hybridization. The four sp3 hybrid orbitals of carbon are directed towards the corners of a regular tetrahedron:
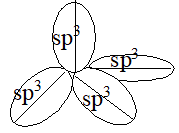
This way the bonding orbitals of carbon are as far apart as far apart as possible. Each hydrogen atom must be located at a corner of this tetrahedron so that each of these hybrid orbitals can overlap most effectively with the spherical s-orbital of a hydrogen atom to form the strongest possible bond. Thus bond angle in methane is 109028′ or 109.50.
2. Source: Methane is the major constituent (up to 97%) of natural gas. It also occurs in the gases from oil-wells. Methane is the principal end product of the anaerobic (without air) decay of plants in swamps and marshes. The gas is set free by the action of bacteria. This method of formation in nature has given rise to the name “marsh-gas” for methane.
Sewage sludge fermented by bacteria yields a gas containing about 70% methane, and this is used as a liquid fuel. Methane also makes about 40% by volume of coal gas. Methane is the dangerous firedamp of the coal mine.
3. Preparation: Methane gas may be synthesized or prepared by the following methods:
i) Striking an electric arc between carbon electrodes in an atmosphere of hydrogen:
ii) Heating a mixture of carbon and reduced nickel at 4750C in the presence of hydrogen:
iii) Passing a mixture of hydrogen and carbon monoxide (or dioxide) over finely divided nickel heated at about 3000C [Sabatier and Senderens method] :
iv) Heating a mixture of anhydrous sodium acetate and sodalime [decarboxylation]
v) Performing reduction of methyliodide with
a) Metals dissolved in suitable solvent: Zn/HCl, Zn/CH3CO2H, Zn/NaOH and Zn–Cu couple/C2H5
b) Lithium aluminium hydride :
c) Triphenyltin hydride
d) Concentrated hydriodic acid in the presence of a small amount of red phosphorus.
e) Via methyl magnesium iodide (a Grignard reagent)
Reduction of formic acid
a) Heating formic acid with hydrogen iodide-red phosphorus under pressure:
b) Heating formic acid with dihydrogen gas under pressure in the presence of a nickel catalyst:
Reduction of formaldehyde
a) Heating formaldehyde with concentrated hydriodic acid in the presence of a small amount of red phosphorus:
b) with amalgamated zinc and concentrated hydrochloric acid [Clemmensen reduction]
c) Heating hydrazone of formaldehyde with sodium ethoxide at 1800C [Wolff-Kshner reduction]
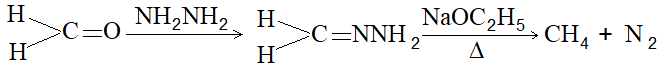
viii) Reduction of methyl alcohol by heating with concentrated hydriodic acid in the presence of a small amount of red phosphorus:
ix) By the action of water on aluminium carbide or beryllium carbide (Hydrolysis) :
x) Methane is obtained in large amounts from natural gas, gas from the oil-wells, and from cracked petroleum. If methane is required in very pure form, it can be separated from the other constituents of natural gas (mostly other alkanes) by fractional distillation. Most of it is consumed as fuel without purification.
4. Physical properties In its physical properties, methane sets the pattern for the other members of the alkene family.
i) Methane is a non-ionic compound. Whether solid, liquid, or gas, its constituent unit is the molecule.
ii) Methane molecule is highly symmetrical. As a result, the polarities of the individual carbon-hydrogen bonds cancel out. Thus, methane is a non-polar covalent compound.
iii) At room temperature, methane is a colourless, odourless and non-poisonous gas.
iv) Melting of solid methane and boiling of liquid methane occur at very low temperatures: p. -1840C, b.p. -1640C/760mm. (Compare these values with the corresponding ones for sodium chloride, a typical ionic compound : m.p. 8010C; b.p. 14130C)
v) In accordance with the rule of thumb that “like dissolves like”, it is only slightly soluble in water [100 mL of water dissolves about 5mL of methane at 200C] but very soluble in non-polar and weakly polar organic solvents such as gasoline, ethanol and ether.
5. Chemical properties In its chemical properties as in its physical properties, methane sets the pattern for the alkane family. Methane is relatively unreactive. It reacts only with highly reactive substances or under very vigorous conditions.
1) Halogenation It is a typical example of a broad class of organic reactions known as substitution -replacement of one or more hydrogen atoms of hydrocarbons by suitable atoms or groups of atoms. Under the influence of diffused sunlight or ultraviolet light or at a temperature of 250 – 4000C a mixture of two gases, methane and chlorine, reacts vigorously to yield hydrogen chloride and chloromethanes:
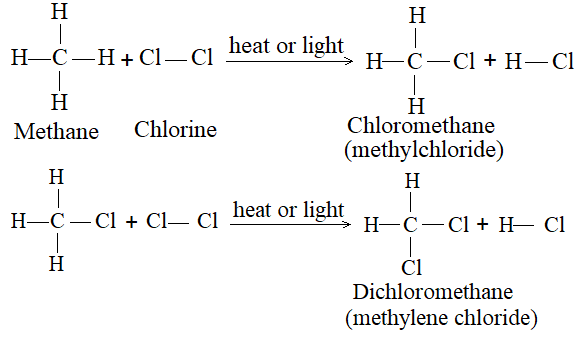

Chlorine has no action on methane in the dark. In bright sunlight the reaction is explosive and hydrogen chloride and carbon are formed:
In diffused sunlight no explosion occurs but a series of substitution reactions takes place. Carbon tetrachloride was once widely us as a non-flammable cleaning agent and as the fluid in certain fire extinguishers, but has been largely replaced by other materials.
ii) Nitration It is the process of substituting a hydrogen atom by a nitro groups, NO2. Methane reacts with the vapours of nitric acid at 400 – 4750C to form nitromethane:
iii) Complete oxidation (Combustion) Methane burns with a non-luminous flame on heating in excess of air or oxygen, forming carbon dioxide and water with the evolution of large amount of heat:
Combustion of methane is the principal reaction taking place during the burning of natural gas Carbon dioxide is the chief of the green house gases, whose accumulation in the stratosphere threatens earth with steadily rising temperature. The other green house gases are methane itself and the chlorofluorocarbons.
Methane explodes violently when mixed with air (or oxygen) and ignited. This is believed to be the cause of explosions in coal mines, where methane is known as fire-damp.
iv) Controlled oxidation Methane on heating with a regulated supply of dioxygen or air under high pressure and at high temperature, in the presence of suitable catalysts undergoes controlled partial oxidation to give a variety of oxidation products:
(a)
(b)
(c)
(d) Ammoxidation (i.e., oxidation in the presence of NH3) of methane is carried out by passing a mixture of methane, air and ammonia over a platinum-rhodium gauze catalyst at 10000C:
It is used to prepare hydrogen cyanide industrially. Other possibilities are:
v) Reaction with steam: Methane reacts with steam at 1273 K (10000C) in the presence of nickel catalyst to form carbon monoxide and dihydrogen gas:
This method is used for industrial preparation of dihydrogen gas.
vi) Pyrolysis Higher alkanes on heating to higher temperature decompose into lower alkenes, alkanes etc. Such a decomposition reaction into smaller fragments by the application of heat is called pyrolysis.
Pyrolysis of alkanes is known as cracking.
Methane, undergoes pyrolysis by heating to 10000C, to produce carbon in a very finely divided state:
Carbon prepared this way is known as carbon black. It can also be obtained through incomplete combustion of methane:
6) Uses:
i) It is consumed as a fuel.
ii) It gives carbon black which is used to make paints and printers’ ink. It is also used in the rubber industry for motor tyres.
iii) It is used as a source of synthesis gas, a mixture of carbon monoxide and hydrogen.
iv) It is used to make dihydrogen (used in the manufacture of ammonia), methanol, formaldehyde and acetylene.
v) It is used as the starting point of many chemicals.
Ethane, C2H6:
The next member of the paraffins after methane is ethane, C2H6. To connect the atoms of this molecule by covalent bonds, we follow the rule: one bond (one pair of electrons) for each hydrogen and four bonds (four pairs of electrons) for each carbon. The result is the following structure:
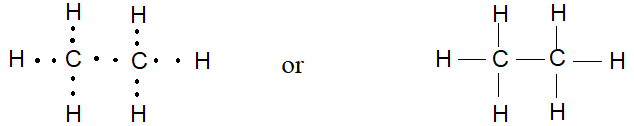
Notice that each carbon is bonded to three hydrogens and to the other carbon.
Since each carbon atom of ethane is bonded to four other atoms, both the carbon atoms in ethane exhibit sp3 hybridization. The four sp3 hybrid orbitals of each carbon are directed towards the corners of a regular tetrahedron. As in the case of methane, the carbon – hydrogen bonds result from overlap of these sp3 hybrid orbitals with the s orbitals of the hydrogens.
The carbon-carbon bond arises from the end-to-end (axial) overlap of two sp3 hybrid orbitals. The carbon-hydrogen and carbon-carbon bonds have the same (general) electron distribution, being cylindrically symmetrical about a line joining the atomic nuclei.
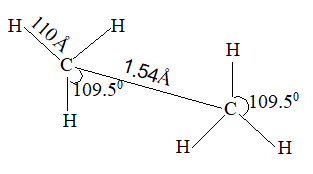
Because of this similarity in shape, the bonds are given the same name, sigma (s) bonds. Thus molecule of ethane has 7 sigma bonds. Electron diffraction and spectroscopic studies have verified, this structure in all respects giving the following measurements for the molecule: HCH or HCC bond angles: 109.50, C-H bond length: 1.10Å, C – C bond length : 1.54 Å and the C-C bond energy 341.11kJ/mol.
With only slight variations, these values are quite characteristic of carbon-hydrogen, carbon-carbon bonds and of carbon bond angles in alkanes.
1. Natural source It occurs with methane in natural gas and the gases from the oil-wells.
2. Preparation Ethane may be prepared or synthesized by the following methods:
i) The best way of preparing pure ethane in large quantity is by the catalytic hydrogenation of ethylene:
, Sabatier-Senderens reduction
ii) Direct reduction of ethyl iodide by the action of
a) dissolved metals : Zn/HCl, Zn/CH3CO2H, Zn/NaOH and Zn-Cu couple / C2H5
b) lithium aluminium hydride:
c) triphenyltinhydride
d) concentrated hydriodic acid in the presence of a small amount of red phosphorus:
iii) The direct reduction of ethyl iodide via a Grignard reagent:
iv) Performing Wurtz reaction with methyl iodide:
v) Reduction of acetic acid by
a) heating acetic acid with hydrogen iodide-red phosphorus under pressure:
b) heating acetic acid with dihydrogen gas under pressure in the presence of a nickel catalyst:
vi) Heating sodium propionate with sodalime (Decarboxylation):
vii) Electrolysis of a concentrated aqueous solution of the sodium or potassium acetate:
viii) Reduction of acetaldehyde by
a) heating acetaldehyde with concentrated hydriodic acid in the presence of a small amount of red phosphorus:
b) amalgamated zinc and concentrated hydrochloric acid [Clemmensen reduction]:
c) heating hydrazone of acetaldehyde with sodium ethoxide at 1800C [Wolff–Kishner reduction].
ix) Reduction of ethyl alcohol by heating with concentrated hydriodic acid in the presence of a small amount of red phosphorus.
3. Physical properties
i) At room temperature, ethane is a colourless, odourless, and non-poisonous gas.
ii) Melting of solid ethane and boiling of liquid ethane also occur at very low temperatures: m.p. -1720C, b.p. -50C.
iii) In agreement with the rule of thumb that “like dissolves like”, it is sparingly soluble in water but readily soluble in organic solvents such as ethanol and ether.
4. Chemical properties
Like methane, ethane also reacts only with highly reactive substances or under very vigorous conditions.
i) Halogenation Ethane reacts with halogens in a similar manner to methane to form substitution products, but relatively a large number of products are possible: firstly due to the presence of six hydrogen atoms in ethane, compared with four in methane, secondly due to the fact that isomerism is possible in the substitution products of ethane (and not in those of methane). For example, two dichloroethanes are possible: .
ii) Nitration Ethane undergoes vapour phase nitration at 4000C to form nitroethane and nitromethane:
iii) Combustion Ethane burns on heating in excess air or oxygen forming carbon dioxide and water with the evolution of large amount of heat:
4) On heating in the absence of air or oxygen, ethane decomposes into ethene and hydrogen:
5. Uses
i) Ethane is mainly used as a fuel.
ii) It is used to prepare ethylene gas.
iii) It is used to prepare chloro (or bromo) ethanes.
Exercise 2:
(i) Knocking sound is produced in the enzinge when the fuel contains
(A) water
(B) lubricating oil
(C) straight-chained hydrocarbons
(D) iso-carbon atoms
(ii) The fraction obtained between temperature 150-3000C during fractional distillation of crude petroleum is
(A) paraffin wax
(B) heavy oil
(C) kerosene
(D) naphtha
(iii) Which of the following compounds has been given an octane number of 100?
(A) n-Hexane
(B) Iso-octane
(C) Neo-pentane
(D) Neo-octane
Exercise 3:
(i) A gaseous hydrocarbon ‘X’ on reaction with bromine in light forms a mixture of two monobromo alkanes and HBr. The hydrocarbon ‘X’ is
(A) C2H6
(B) C3H6
(C) C3H8
(D) C4H10
(ii) 2.44g of methyl iodided was completely converted into methyl magnesium iodided and the product was decomposed by excess of ethanol. The volume of the gaseous hydrocarbon produced at NTP will be
(A) 22.4 litre
(B) 22400 mL
(C) 0.448 litre
(D) 0.224 litre
(iii) In the complete combustion of CnH2n+2, the number of oxygen moles required is
(A)
(B)
(C)
(D)
ALKENES
The alkenes can be obtained from alkanes by loss of hydrogen in the cracking process. They can be converted into alkanes by addition of hydrogen. Since alkenes (evidently) contain less than the maximum quantity of hydrogen, they are referred to as unsaturated hydrocarbons. They contain just one double bond and thus have the general formula CnH2n. The double bond of alkenes is sometimes known as the ‘olefinic bond’ or ‘ethylenic bond’. Alkenes are also known as olefins (oil-forming). This name arose from the fact that the simplest member of the alkene family, ethylene, was called ‘olefiant gas’ (oil-forming gas), since it formed oily liquids on treatment with halogens (chlorine or bromine).
Structure of the carbon-carbon double bond
Due to ready transformation of ethylene into ethane, one expects certain structural similarities between the two compounds.
It we connect the two carbon atoms of ethylene, C2H4, by a covalent bond and then attach two hydrogen atoms to each carbon atom:
we find that each carbon atom possesses only six electrons in its valence shell, instead of the much needed eight electrons. Moreover, the entire molecule needs an additional pair of electrons to remain neutral. These two problems can be solved if the two carbon atoms share two pairs of electrons i.e., the carbon atoms are joined by a double bond.
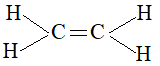
The carbon-carbon bond is the distinguishing feature of the alkene structure.
Whenever carbon is bonded with three other atoms, it makes use of three identical sp2 hybrid orbitals (formed by the mixing of one s and two p orbitals). These three sp2 hybrid orbitals always lie in one plane (that of the carbon nucleus) and are always directed torward the corners of an equilateral triangle. Thus, the angle between any pair of sp2 hybrid orbitals is 1200.
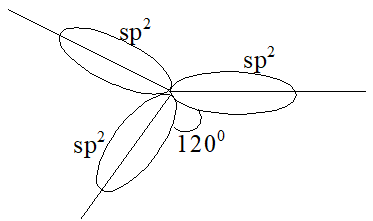
This trigonal arrangement allows the three sp2 hybrid orbitals to be as far apart as possible, and gives three trigonal bonds.
To permit maximum overlap of orbitals, the two carbons and four hydrogens of ethylene should be arranged as shown below:
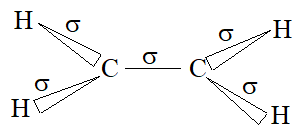
Each carbon atom is lying at the centre of a triangle whose corners are occupied by the two hydrogen atoms and the other, by carbon atom. Thus every bond angle is 1200. All these bonds are cylindrically symmetrical about a line joining the nuclei, and are thus called the sigma (s) bonds. To form the sp2 hybrid orbitals, each carbon atom has used only two of its three p orbitals. The remaining pure p orbitals consists of two equal lobes, one lying above and the other lying below the plane of the three sp2 orbitals. It is occupied by a single electron. If the p-orbital of one carbon atom overlaps with the p orbital of another carbon atom, the two electrons pair up and an additional bond is formed between the two carbons.
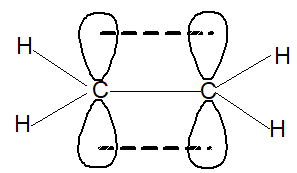
This new bond formed by the sideways overlap of p orbitals is called a pi (p) bond. It is differently shaped. It consists of two parts, one half of the electron cloud lies above the plane of the atoms, and another half of the electron cloud lies below.
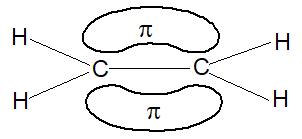
Because of poor sideways overlap between the two p orbitals, the p bond is weaker than the carbon-carbon s bond. Since sideways overlap of p-orbitals can occur only when all six atoms (two carbons and four hydrogens) lie in the same plane, ethylene is a flat molecule. The carbon-carbon double bond in alkenes thus consists of one strong sigma (s) bond (bond enthylpy about 397 kJ/mol) formed by head-on overlapping of sp2 hybridised orbitals and one weak pi (p) bond (bond enthalpy about 284 kJ/mol) obtained by lateral or sideways overlapping of the two p orbitals of the two carbon atoms.
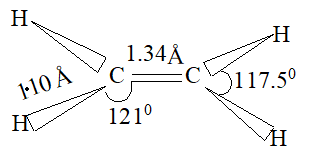
The total bond energy of the double bond (681 kJ mol–1 or 146 kcal mol–1) is greater than that of the carbon-carbon single bond of ethane (384 kJ mol–1 or 88 kcal mol–1). Since the doubly bonded carbon atoms are held more tightly together, the carbon-carbon distance in ethylene is less than the carbon-carbon distance in ethane. In other words, the carbon-carbon double bond (134pm) is shorter than the carbon-carbon single bond (154 pm).
Electron diffraction and spectroscopic, studies show ethylene to be a flat molecule, with bond angles very close to 1200. The C-C distance is 1.34Å as compared with the C-C distance of 1.53Å in ethane:
The bond in ethylene has been evaluated to have a strength of about 95 kcal. It is stronger than the bond in ethane because it is formed by overlap of sp2 hybrid orbitals. On this basis, one can estimate the strength of the bond to be 51 kcal.
Nomenclature
For nomenclature of alkenes refer to phase II’s study package summary. The longest continuous chain of carbon atoms containing the double bond is selected. Numbering of the chain is done from the end which is nearer to the double bond. The suffix ‘ene’ replaces ‘ane’ of alkanes.
First member of alkene series is CH2 (substituting n as 1 in CnH2n) known as methene or methylene but it has a very short life. Thus, the first stable member of alkene series is C2H4.
Illustration 5: Give structures or write IUPAC names of the following compounds
a) 4–Ethyl–2, 6–dimethyldec–4–ene
b)
c) 1, 3, 5, 7 – Octatetraene
d)
Solution:
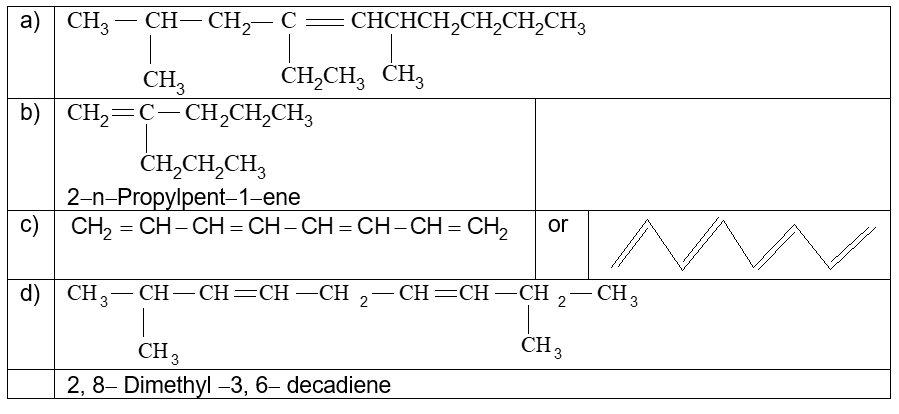
Illustration 6: Count the number of sigma () and pi () bonds in the above compounds (a–d).
Solution:
a) bonds : 41, bond : 1
b) bonds : 23, bond : 1
c) bonds : 17, bond : 4
d) bonds : 33, bond : 2
Isomerism
Alkenes exhibit both structural and stereoisomerism.
Structural isomerism As in alkanes, there is only one possible structure for ethene (C2H4) and propene (C3H6). Going on to the butylenes, C4H8, we notice that there are more than one possible arrangements:
a) We may have either a straight-chain skeleton as in n-butane, or a branched-chain skeleton as in iso-butane.
b) For the straight-chain skeleton there are two possible arrangements that differ in the position of the double bond in the chain.
This leads to a total of three different structures
Structures I and III, and II and III are examples of chain isomers while structures I and II are examples of position isomers.
Illustration 7: Work out all possible structurally isomeric alkenes corresponding to C5H10 and also give their IUPAC names.
Solution: Straight chain skeleton:
a)
b)
c)
d)
e)
The lack of rotation around the carbon-carbon double bond has an important chemical consequence. Consider a disubstituted alkene such as but-2-ene.
Disubstituted means that the two substituents other than hydrogen are bonded to the double-bond carbons.
The two methyl groups in but-2-ene can be either on the same side of the double bond or on the opposite sides:
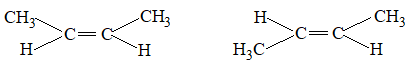
Geometrical Isomerism
Since bond rotation can’t occur, the two but-2-enes can’t spontaneously interconvert. They are different isolable compounds called geometrical isomers or cis-trans stereoisomers. For further details refer to study package of phase II.
Illustration 8: Which of the following compounds will exhibit geometrical (or cis-trans) isomerism.
a)
b)
c)
d)
Solution:
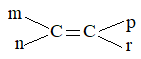
m n and p r
Thus structures (a) and (b) will exhibit geometrical isomerism. The structures (c) and (d) fail to do so because two identical atoms (for c) and groups (for d) are attached to one of the doubly bonded carbon atoms.
Illustration 9: Draw geometrical (or cis–and trans–) isomers of the following structures, and give their IUPAC names.
a)
b) CHCl = CHCl
Solution:

General Methods of Preparation of the Alkenes
The smaller alkenes i.e., alkenes containing up to four carbon atoms (ethylene, propylene, and butylenes) can be obtained in pure form from the petroleum industry by cracking. Pure samples of more complicated alkenes must be prepared by methods like those discussed below.
There are basically three types of methods to prepare alkenes:
a) Introduction of a carbon-carbon double bond into a molecule containing only single bonds. It essentially involves the elimination of identical or different atoms or groups from two adjacent carbons:

X and Y may be same or different.
b) Generating a carbon–carbon double bond from a carbon–carbon triple bond by an addition reaction:

c) Conversion of one alkene into another. It essentially involves the isomerisation.
1) Dehydrohalogenation of alkyl halides
When alkyl halides (R-X) are heated with concentrated alcoholic solution of potassium hydroxide, alkenes are obtained along with potassium halide and water:
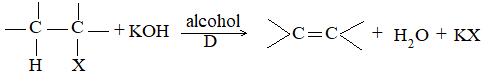
Ease of dehydrohalogenation :
of alkyl halides :
Examples
This reaction, known as dehydrohalogenation i.e., elimination of the elements of hydrogen halide, is a typical example of the 1, 2- or – elimination reaction, in which two atoms or groups are lost from neighboring atoms to form a double bond.
Other elimination reactions (, , etc) are also known, but these are much less common. Dehydrohalogenation involves removal of the halogen atom (from – carbon) and of a hydrogen atom from a carbon (b-carbon) adjacent to the one losing the halogen. The reagent required is a strong base (like potassium ethoxide) whose function is to abstract the hydrogen as a proton.
The products of the reaction clearly show that halogen leaves the alkyl halide as halide ion, taking the shared electron pair along and hydrogen is abstracted by the base (ethoxide ion) as a proton, leaving the shared electron pair behind. It is this electron pair that is used up to form the additional bond (the bond) between the carbon atoms.
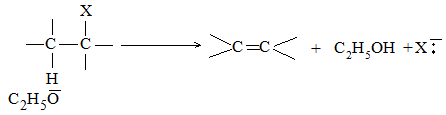
In some cases, dehydrohalogenation yields a mixture of alkenes. This is due to the fact that an alkene is expected corresponding to the loss of any one of the possible -hydrogens. n-Butyl halide yields only one alkene, but-1-ene, as it can lose hydrogen only from C-2:
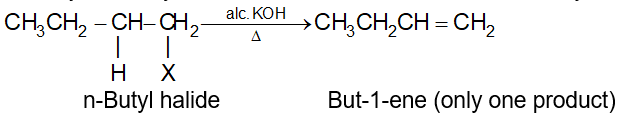
On the other hand, sec-Butyl bromide yields two alkenes, but-1-ene and but-2-ene corresponding to the loss of b-hydrogen either from C-1 or from C-3:
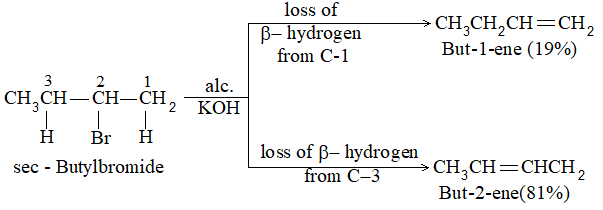
Notice that the preferred (or major) product, but-2-ene is a disubstituted alkene i.e., there are two alkyl groups (two methyl groups) attached to the doubly bonded carbons of but-2-ene. The minor product, but-1-ene is a monosubstituted alkene i.e. there is only one alkyl group (one ethyl group) attached to the doubly bonded carbons:
predominates over
The following examples also show that a disubstituted alkene predominates over a monosubstituted alkene, and a trisubstituted alkene predominates over a disubstituted alkene:
disubstituted trisubstituted
alkene (29%) alkene (71%)
This pattern was first observed by the Russian chemist Alexander Saytzeff who formulated (in 1875) an empirical rule which may be stated in two ways:
In dehydrohalogenation (and also dehydration of alcohols) the predominant product is the most substituted alkene i.e., the one carrying the maximum number of alkyl substituents.
or
In dehydrohalogenation (and also dehydration of alcohols) -hydrogen is eliminated preferentially from that b- carbon atom which is joined to the least number of hydrogen atoms.
Since the amount of each isomer formed depends on its rate of formation, we can conclude that the alkene with the greater number of alkyl substituents is the predominant product because it is formed faster than alternative alkenes. Thus, Saytzeff rule gives us a sequence showing the relative rates of formation of alkenes.
Ease of formation of alkenes:
Notice that the stability of alkenes also follows the exact sequence:
Stability of alkenes:
Now we can redefine the Saytzeff’s rule to state:
In dehydrohalogenation, the more substituted alkene, being the more stable, is formed faster. Predominant formation of the more stable isomer is called Saytzeff orientation.
Kinetics of elimination
With a concentrated alcoholic solution of potassium hydroxide, dehydrohalogenation follows second-order kinetics:
Rate = k [RX] [base]
i.e., the rate of formation of alkene depends upon the concentration of alkyl halide and base. This behaviour is observed for all classes of alkyl halides.
If one moves along a series of substrates, 10RX to 20 RX to 30RX, and if one reduces the concentration or the strength of the base, the dehydrohalogenation begins to follow first –order kinetics :
Rate = k [RX]
i.e., the rate of alkene formation (or the rate of elimination) depends only upon the concentration of alkyl halide, and is independent of the strength or concentration of base. This first -order kinetics is observed only with secondary or tertiary halides, and in solutions containing either a weak base or having low concentration of base.
To explain the two different kinetics, Hughes and Ingold proposed two different mechanisms for the dehydrohalogenation of alkyl halides. They are named E1 (Elimination through first order kinetics) and E2 (Elimination through second order kinetics or biomolecular elimination).
2) Dehydration of monohydric alcohols
A monohydric alcohol is converted into an alkene by dehydration: 1, 2- or – elimination of a molecule of water:
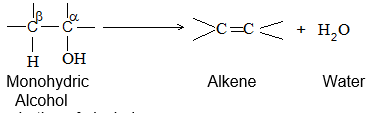
Ease of dehydration of alcohols:
30ROH > 20ROH > 10ROH
Dehydration may be effected by heat alone (400-8000C), but in the presence of a suitable catalyst lower temperatures are required. Unlike dehydrohalogenation (of alkyl halides) that is catalyzed (or promoted) by a base, the dehydration of alcohols is catalyzed by an acid.
Thus, dehydration requires the presence of an acid and the application of heat. It is usually carried out in either of two ways:
i) by heating the alcohol with a Brønsted acid such as sulphuric acid or phosphoric acid. Primary alcohols are dehydrated by the action of concentrated sulphuric acid at about 160-1700. Dehydration of secondary and tertiary alcohols is best carried out by using boiling dilute sulphuric acid, since the alkenes produced from these alcohols (particularly tertiary alcohols) tend to polymerise under the influence of the concentrated acid.
ii) by passing the alcohol vapor over heated alumina (Al2O3) which functions either as a Lewis acid or through -OH groups on its surface, as a Lowry -Brønsted 30 alcohols are dehydrated at about 1500C, 20 -alcohols at 2500C and 10 alcohols at 3500C.
The yields of alkenes from secondary and tertiary alcohols are very good.
Acid-catalysed dehydration of primary alcohols usually yields 1-alkenes, but 20 – and 30-alcohols yield mixtures of alkenes due to removal of a hydrogen (as proton) from either adjacent carbon atom (to the COH group) and also due to rearrangements undergone by the different carbonium ion intermediates.
Rearrangements may be avoided by dehydrating the alcohol over heated alumina and isomerisation is suppressed by the addition of a small amount of pyridine to alumina.
Examples:
Orientation of elimination is strictly Saytzeff
Where more than one b-carbon is available then hydrogen attached to the b- carbon atom joined to the least number of hydrogen atoms is eliminated most easily. For example:
Thus the predominant product is the more substituted and hence the more stable one.
3) Dehalogenation of dihalides
Dihalogen derivatives of the alkanes undergo dehalogenation (i.e. removal of halogen) on treatment with zinc dust and methyl alcohol to produce the corresponding alkenes:
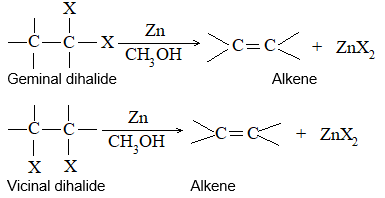
Examples
A probable mechanism is
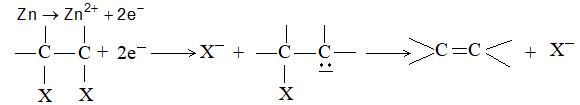
Neither method is useful for making alkenes, since the required dihalogen compounds are not easily accessible. Dehalogenation of vicinal (Latin: vicinalis, neighboring) dihalides is severally limited by the fact that these dihalides are themselves prepared from the alkenes.
However it is sometimes useful for purifying alkenes or for ‘protecting’ a double bond:
Usually oxidizing agents attack the double bond in allyl alcohol as well as the alcoholic group. However, oxidation may be carried out by ‘protecting’ the double bond by bromination and then oxidizing and debrominating with zinc dust in methanolic solution:
Sodium iodide may be used instead of zinc dust:
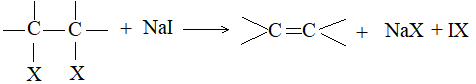
Mechanism
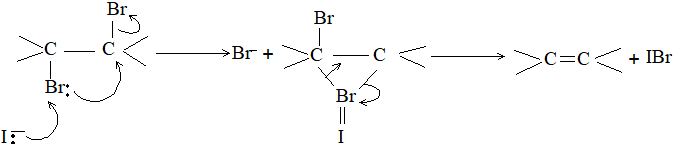
Other means of removing the two halogen atoms to regenerate the alkene involve treatment with chromous chloride, chromus sulphate, or sodium trimethoxyborohydride. Both addition (to the alkene) and elimination (from the dihalide) of the two halogen atoms are predominantly trans i.e. setereoselective in accordance with the formation of a bridged intermediate. Consequently the configuration of the protected alkene remains unchanged.
4) Heating a quaternary ammonium hydroxide
For predicting the major alkene, here we use Hoffman elimination rule which is just opposite to that of Saytzeff rule.
5) The Wittig reaction
It is the reaction of alkylidenephosphoranes (also called ylides) with carbonyl compounds to form alkenes:
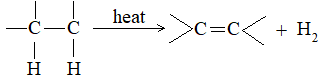
6) Cracking process
Alkenes containing up to four carbon atoms (ethene, propene, butenes) are prepared by the cracking of petroleum. In the cracking process, the atoms eliminated are both hydrogen atoms:
For the production of the lower alkenes the most suitable starting material is gas oil, whereas for the higher alkenes it is best to use paraffin wax or Fisher–Tropsch wax.
The lower alkenes (C2-C5) can also be prepared by the catalytic dehydrogenation of saturated hydrocarbons, the most satisfactory catalyst being chromium oxide-alumina.
7) Reduction of alkynes
Alkynes add on hydrogen in the presence of a catalyst, the reaction proceeds in two stages:
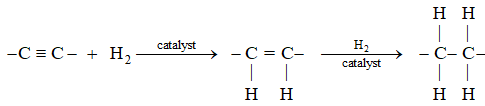
Many catalysts affect hydrogenation of a triple bond. By using suitable conditions, it is possible to isolate the intermediate alkene.
Reduction of a non-terminal alkyne to the double-bond stage can yield either a cis alkene or a trans alkene. Just which isomer predominates depends upon the choice of reducing agent.
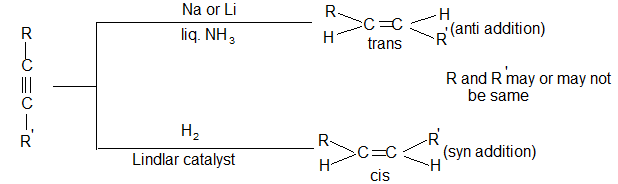
Predominantly trans alkene is obtained when a dialkylacetylene is reduced with sodium or lithium in liquid ammonia. Trans-alkene is also produced by reduction with lithium aluminium hydride.
Almost entirely cis alkene is obtained by hydrogenation of dialkylacetylene with Lindlar’s catalyst which consists of a Pd-CaCO3 catalyst partially poisoned with lead -acetate. A better catalyst is Pd-BaSO4 partially poisoned with quinoline or palladised charcoal partially deactivated with quinoline. Cis-alkene is also the predominant product when reduction is carried out with diborane (B2H6) or with di-isobutylaluminium hydride.
Physical properties of alkenes
1) Physical state: The first three members containing two to four carbon atoms are gases, the next thirteen containing five to seventeen carbon atoms are liquids and the higher ones containing eighteen carbon atoms onwards are solids at room temperature.
2) Boiling points and melting points: The boiling point of an alkene is very nearly the same as that of the alkane with the corresponding carbon skeleton. The boiling point rises with increasing carbon number, as with alkanes. The boiling point rise is 20-30 degrees for every added carbon, except for the very small homologs. As with alkanes, branching lowers the boiling point of alkenes.
Like alkanes, alkenes are only weakly polar. Since the loosely held p electrons of the carbon-carbon double bond are easily pulled or pused, dipole moments are relatively larger than for alkanes. However, they are still small. For example:

Compare the dipole moments for propene and but-1-ene with the dipole moment of 1.83D for methyl chloride.
Cis form of alkene is found to be more polar than the trans form. For example, cis-but-2-ene, with two methyl groups on one side of double bond and two hydrogens on the other, has a small dipole moment of 0.33 Debye whereas in trans-but-2-ene, with one methyl and one hydrogen on each side of the molecule, the dipole moments cancel out. Consequently, cis form is polar while trans form is non polar.
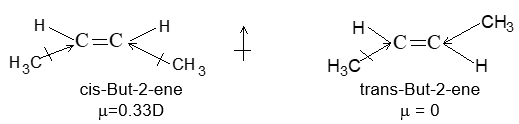
Because of its higher polarity the cis isomer has usually the higher boiling point for a pair of geometrical isomers. However, because of its lower symmetry, the cis isomer fits more poorly into a crystal lattice and thus usually has the lower melting point.
For alkenes containing elements whose electronegativities differ widely from that of carbon, the differences in polarity, and hence the differences in boiling point and melting point are quite higher:

3) Solubility: They are almost insoluble in water but quite soluble in non-polar or weakly solvents polar like benzene, ether, chloroform, or ligroin.
4) Density: They are less dense than water.
Chemical properties of alkenes
The carbon-carbon double bond, the characteristic feature of the alkene structure, is the functional group of alkenes and hence determines the characteristic reactions that alkenes undergo. These reactions are of two types:
first, those reactions that take place at the double bond itself, next, those reactions that do not take place at the double bond but occur at the alkyl part of the alkene.
The double bond consists of a strong sigma (s) bond and a weak pi (p) bond. The typical reactions of the double bond are those where the pi bond is broken and two strong sigma bonds are formed in its place:
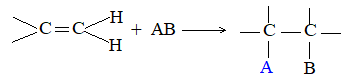
A reaction involving the combination of two molecules to form a single molecule of product is called an addition reaction. Addition reactions are essentially the characteristic reactions of compounds containing multiply bonded atoms. Owing to the presence of a double bond, the alkenes undergo a large number of addition reactions.
In carbon-carbon double bond there is a cloud of electrons above and below the plane of the atoms.
Thus, these electrons are less involved than the electrons in holding together the carbon nuclei. Consequently, they are themselves held less tightly. These loosely held electrons are easily available to any reagent that is in need of electrons. Thus, in most of its reactions the carbon- carbon double bond serves as a rich source of loosely held pi () electrons and hence acts as a Lewis base. The compounds with which carbon-carbon double bond of alkenes reacts are evidently deficient in electrons and hence act as Lewis acids. These acidic reagents (Lewis acids) that are requiring a pair of electrons are called electrophilic reagents (Greek: electrophiles, electron-loving).
Thus the characteristic reaction of an alkene is electrophilic addition (i.e. addition of acidic reagents). Alkenes also undergo free-radical addition where the reagents, the free radicals, need just an electron.
Except ethene, alkenes contain not only the carbon-carbon double bond but also alkyl groups having essentially the alkane structure. Therefore, alkenes under special conditions may undergo the free-radical substitution reactions typical of alkanes.
General mechanism of electrophilic addition reaction
When addition occurs, the trigonal planar arrangement in the alkene changes into tetrahedral. In the first step, alkene’s addition reactions involve the attack of an electrophile. This is the rate-controlling step. In the second step, the intermediate, a carbonium ion, reacts rapidly with a nucleophile. If E and Nu, describe the electrophilic and nucleophilic fragments of the addendum E-Nu, the addition mechanism may be generalized as follows:
Step 1 :
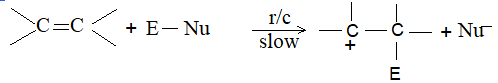
Step 2:
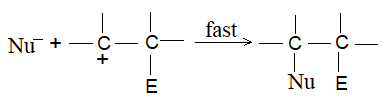
1) Addition of dihydrogen
Alkenes add up one molecule of dihydrogen gas in the presence of a suitable catalyst at the carbon-carbon double bond to form alkanes of the same carbon skeleton. The process called catalytic hydrogenation is of two kinds: (i) heterogeneous (two-phase) and (ii) homogeneous (one-phase).
Heterogeneous hydrogenation is the classical method. A solution of alkene is shaken with dihydrogen gas under pressure in the presence of a suitable catalyst. Finely divided platinum and palladium are effective at room temperature; nickel on a support (Sabatier–senderens reduction) requires a temperature between 2000C and 3000C; Raney nickel is effective at room temperature and atmospheric pressure.
Example
Reaction takes place rapidly and smoothly. When the reaction is complete, the solution of the saturated product is simply filtered from the insoluble catalyst. One molecule of dihydrogen gas is consumed for each double bond present in the unsaturated compound.
In homogeneous hydrogenation the catalysts are organic complexes of transition metals like rhodium or iridium, for example, Wilkinson’s catalyst . They are soluble in organic solvents, thus, hydrogenation takes place in a single phase, the solution.
Hydrogenation is frequently used as an analytical tool as it can tell us the number of double bonds in a compound because the reaction is quantitative and the volume of dihydrogen gas can be easily measured. Hydrogenation is exothermic as the two sigma bonds (C-H) formed are stronger than the sigma bond (H-H) and bond broken. The amount of heat energy released when one mole of an unsaturated compound in hydrogenated is called the heat of hydrogenation. The heat of hydrogenation of nearly every alkene is fairly close to a value of 30 kcal for each double bond in the compound. Heat of hydrogenation can be used to compare the stabilities of different alkenes which produce the same alkane on hydrogenation. Since the hydrogenation is exothermic, the smaller H is (numerically), the more stable is the alkene to its parent alkane. The following examples are typical:
– 115.5 kJ mol–1 -119.7 kJ mol–1 -126.8 kJ mol–1
– 27.6 kcal mol–1 – 28.6 kcal mol–1 -30.3 kcal mol–1
Heat of hydrogenation for is -118.8 kJ/mol
-118.8 kJ/mol
Each set of isomeric alkenes yields the same alkane. In each case, the greater the number of alkyl substituents attached to the doubly bonded carbon atoms, the more stable the alkene. For a pair of geometrical isomers, the trans form is more stable than the cis form.
Order of stability of alkenes
Stabilities of alkenes may also be compared by the determination of their heats of combustion. For example
In this case all four butanes may be compared, since all give the same products on combustion, namely, 4CO2 + 4H2O.
The rate of hydrogenation of olefinic bonds (at room temperature and atmospheric pressure) is :
When the alkene is of the type R2C = CR2 or R2C = CHR, then hydrogenation at room temperature and atmospheric pressure is difficult. The results are in accordance with the fact that the less hindered side of an unsaturated molecule is adsorbed on the metal surface.
The olefinic bond is not reduced by metal and acid, sodium and ethanol, lithium aluminium hydride unless it is a, b with respect to certain reducible groups e.g. > C = O.
Birch reduction is used to reduce a terminal double bond by sodium in liquid NH3 in the presence of an alcohol (CH3OH or C2H5OH):
2) Addition of halogens Alkenes readily add up chlorine or bromine to form saturated compounds containing two atoms of halogen attached to adjacent carbons. Iodine usually fails to react under normal conditions.
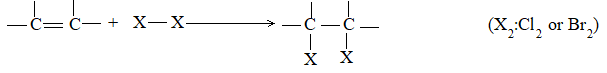
This addition reaction is the best method of preparing vicinal dihalides. The reaction is usually carried out simply by mixing together the two reactants in an inert solvent such as carbon tetrachloride. The addition reaction proceeds rapidly in solution at room temperature in the absence of light or peroxides. We should avoid higher temperatures and undue exposure to light as well as the presence of excess halogen, since under these conditions substitution becomes an important side reaction.
Examples :
Addition of bromine is used as a test for unsaturation.
The reddish orange color of the solution of bromine in carbon tetrachloride is rapidly discharged when bromine adds up to the carbon-carbon double bond:
The addition of halogens to alkenes is an electrophilic addition reaction involving two steps. The first step involves the formation of a cyclic halonium ion:
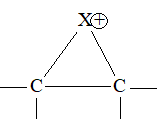
3) Addition of hydrogen halides Alkenes add up hydrogen halides (HCl, HBr and HI) to form the corresponding alkyl halides:
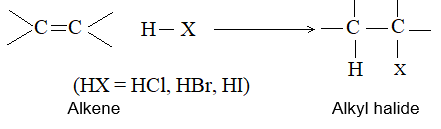
The reaction is frequently carried out by bubbling the dry gaseous hydrogen halide directly into the alkene (using the alkene itself as the solvent). Sometimes these additions are carried out by dissolving both the polar hydrogen halide and the non-polar or weakly polar alkene in a moderately polar solvent such as acetic acid or methylene chloride (CH2Cl2).
Aqueous solutions of the hydrogen halides are not used, to avoid, the addition of water to the alkene. The order of reactivity of the addition of hydrogen halides is:
This is also the order of acid strength. The reactivity of HF is so low that the addition of hydrogen fluoride is effected only under pressure.
Examples
The addition of HX to an unsymmetrical alkene could take place in two different ways to yield two different products, depending upon the orientation of addition (i.e. depending upon which carbon atoms get attached to the hydrogen and halogen).
While examining a large number of such additions, the Russian chemist Vladimir Markovnikov observed that whenever two isomeric products are possible, one of them usually predominates. Thus during addition of HBr to propene, the main product is 2-bromopropane, and when isobutylene reacts with HI, the main product is tert-butyl iodide. He pointed out (1869–1870) that the preferred orientation of addition follows a pattern which can be summarized as Markovnikov’s rule: In the addition of an unsymmetrical reagent (such as HX) to an unsymmetrical alkene (such as propene) the positive part of the reagent (such as the hydrogen atom of the HX) adds to the carbon atom of the double bond that already has the greater number of hydrogen atoms. Alternatively, it can be stated as: the negative part of the addendum attaches itself to the carbon atom of the double bond that is joined to the least number of hydrogen atoms.
Thus in the addition of HBr to propene, the hydrogen atom attaches itself to the doubly bonded carbon carrying two hydrogen atoms rather than to the doubly bonded carbon carrying just one hydrogen:
Similarly, in the addition of HI to isobutylene, the hydrogen adds to the carbon bearing two hydrogens rather than to the carbon bearing one:
Reactions that illustrate (or follow) Markovnikov’s rule are said to be Markovnikov additions.
Illustration 10: Using Markovnikov’s rule, correctly predict the principal product of the following reactions.
a) But-1-ene + HI
b) Vinyl chloride + HI
c) But-2-ene + HI
d) 2-Methyl-but-2-ene + HI
e) Pent-2-ene + HI
Solution:
a)
b)
c)
2-Butene is a symmetrical alkene, thus, only one product is obtained.
d)
e)
In pent-2-ene, each of the doubly bonded carbons holds one hydrogen, thus, neither product should predominate in accordance with the rule. Roughly equal amounts of the two isomers are obtained.
A mechanism for addition of a hydrogen halide
Markovnikov’s rule is empirical but could be explained theoretically on the basis that the addition occurs by a polar mechanism. As with halogens, the addition of hydrogen halides is an electrophilic addition reaction involving the following two steps:
Step 1 Addition of proton – The electrons of the alkene form a bond with a proton from HX to form a carbocation and a halide ion:
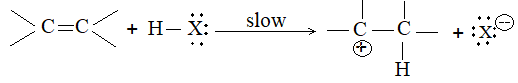
Step 2 Addition of halide ion – The halide ion reacts with the carbocation by donating an electron pair; the result is an alkyl halide:
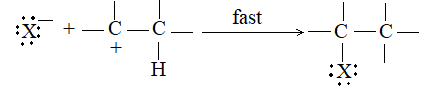
The addition is predominantly trans on account of the formation of a bridged carbonium ion:
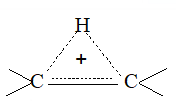
Step 1 has a high energy of activation and takes place slowly while step 2 has a very low energy of activation and takes place very rapidly. Thus, step 1 is the rate-controlling step.
Theoretical explanation of Markovnikov’s rule
Any factor that lowers the activation energy of the rate-determining step will increase the rate of addition of hydrogen halides. Hence, the more stable the carbocation, the faster will be the addition. Since the amount of each isomer formed is directly proportional to its rate of formation, the faster reaction will lead to the formation of the main product.
If an unsymmetrical alkene, such as propene, undergoes addition of a hydrogen halide, such as hydrogen bromide, then step 1 leads to two different carbocations:
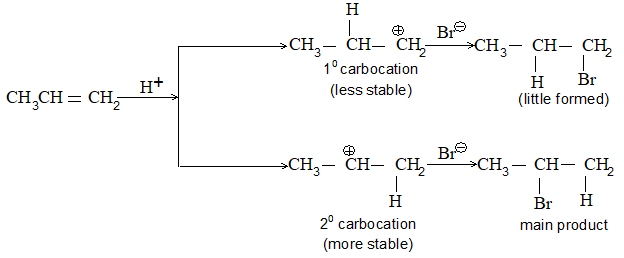
However, these two carbocations are not of equal stability. The secondary carbocation is more stable than the primary carbocation and hence is formed faster. The chief product of the reaction is thus 2-bromopropane because it is formed by the attack of bromide ion on the more stable secondary carbocation which is formed preferentially in the first step.
Since the order of stabilities of carbocations is the rate of addition of HX will be:
Because carbocations are formed in the addition of HX to an alkane, rearrangements may occur in certain cases, according to the nature of the carbocation, when the carbocation initially formed can rearrange to a more stable one.
Illustration 11: In view of the ionic addition of hydrogen halides to alkenes, write the modern statement of Markovnikov’s rule?
Solution: Because step 1 determines the overall orientation of the reaction, the modern statement of the rule is:
In the ionic addition of an unsymmetrical reagent to a double bond, the positive portion of the addendum attaches itself to a carbon atom of the double bond so as to yield the more stable carbocation as an intermediate.
Illustration 12: Predict the outcome of the addition of ICl and BrCl to propene.
Solution: Chlorine being more electronegative than either iodine or bromine, is the negative part of the addendum and thus adds in the second step:
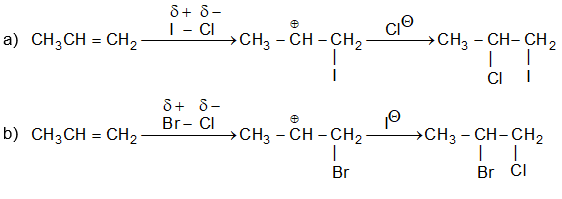
Reactions such as the Markovnikov’s addition of hydrogen halides are described as regioselective reactions. Regio (pronounced as “reejio”) comes from the Latin word regionem meaning direction. Reactions that, from the view point of orientation, yield almost or nearly almost one of several possible isomeric products are called regioselective.
or
When a reaction that can potentially give two or more structurally isomeric products actually produces only one (or a predominance of one), the reaction is said to be regioselective.
Peroxide effect – an exception to Markovnikov’s rule:
Addition of hydrogen chloride and hydrogen iodide to unsymmetrical alkenes always follows Markovnikov’s rule. Until 1933, the orientation of the addition of hydrogen bromide to unsymmetrical alkenes was highly confused. Some chemists reported that addition of hydrogen bromide yields a product in agreement with Markovnikov’s rule, others reported a product against Markovnikov’s rule and still others, said, a mixture of both products. This chemical chaos (or mystery) was solved in 1933 by M.S. Kharasch and F.R. Mayo. They discovered that the orientation of addition of hydrogen bromide to the carbon-carbon double bond of an unsymmetrical alkene is determined completely by the absence or presence of peroxides or hydroperoxides.
Organic peroxides or hydroperoxides are compounds containing the -O-O- linkage.
![]()
An organic peroxide An organic hydroperoxide
They are found, usually in very small amounts, as impurities, in many organic compounds including alkenes, slowly formed by the action of air or oxygen. However, certain peroxides such as tert-butyl peroxide, [(CH3)3CO]2 or benzoyl peroxide, are deliberately synthesized and used as reagents.
Kharasch and Mayo found that the presence of oxygen or peroxides and hydroperoxides that are formed when the unsymmetrical alkene stands exposed to the air, or added peroxides such as benzoyl peroxide, causes the addition of HBr to take place in the direction exactly opposite to that predicted by Markovnikov’s rule. On the other hand, in the absence of peroxides (i.e. if one carefully excludes peroxides from the reaction system), or if one adds certain inhibitors (i.e. compounds that would ‘trap” radicals) for example, hydroquinone or diphenylamine – the addition of HBr to unsymmetrical alkenes follows Markovnikov’s rule:
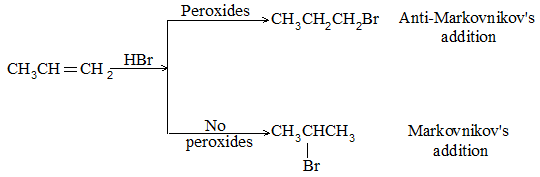
This departure from the rule is known as the ‘abnormal’ reaction and this reversal of the orientation of addition caused by the presence of peroxides is known as the peroxide effect.
Hydrogen fluoride, hydrogen chloride and hydrogen iodide do not undergo anti-Markovnikov’s addition even when peroxides are present.
Mechanism for the anti-Markovnikov addition:
Addition of HBr is ‘abnormally’ affected photochemically as well as by peroxide. The mechanism of the anti-Markovnikov addition of hydrogen bromide is a free-radical chain reaction initiated by peroxides or by light.
Step1 Chain initiation – Heat brings about homolytic cleavage of the weak oxygen-oxygen bond of organic peroxide:
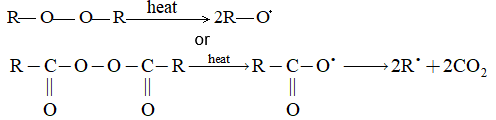
Step 2: Chain propagation – The alkoxy radical (or alkyl radical) abstracts a hydrogen atom from H-Br, producing a bromine atom.
or
Step 3 Chain propagation – A bromine atom adds to the double bond to produce the more stable secondary (20) free radical:
Step 4 Chain propagation – The secondary free radical abstracts a hydrogen atom from H-Br. This leads to the product formation and regenerates a bromine atom.
Then repetitions of steps 3 and 4 lead to a chain reaction.
In the photochemical addition, the bromine atom is produced by absorption of a quantum of light.
For alkyl halide synthesis, polar, protic solvents favour Markovnikov’s addition:
While non-polar solvents or exposure to light or presence of peroxides favour anti- Markovnikov’s addition:
Illustration 13: Find the products obtained by addition of HBr to hex-1-ene
a) in the presence of peroxide
b) in the absence of peroxide
Also write their IUPAC names.
Solution:
a) In the presence of peroxides, the particle that attacks the double bond first is the larger bromine atom. It attaches itself to the less hindered carbon atom by a radical mechanism to form the more stable free radical. The result is anti- Markovnikov’s addition.
b) In the absence of peroxides, the particle that attacks the double bond first is a proton (Because, a proton is small, steric effects are not important). It attaches itself to a carbon atom by an ionic mechanism to form the most stable carbocation. The result is Markovnikov’s addition:
4) Addition of sulphuric acid: On treatment with cold concentrated sulphuric acid, alkenes (unlike alkanes) dissolve because they react with sulphuric acid by addition to form compounds of the general formula ROSO3H or RHSO4, known as alkyl hydrogen sulphates. These products are formed by a mechanism which is similar to that for the addition of HX:
Step 1 The alkene donates a pair of p electrons to a proton from sulphuric acid to form a carbocation:
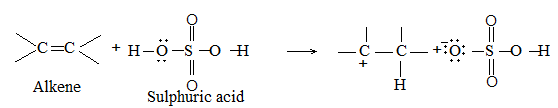
Step 2 The carbocation reacts with a hydrogen sulphate ion to form an alkyl hydrogen sulphate.
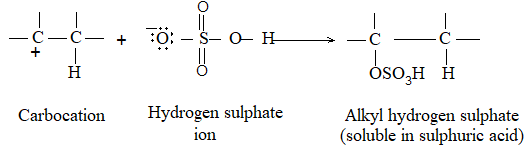
Alkyl hydrogen sulphates are formed by addition of hydrogen to one carbon of the double bond and bisulphate ion to the other.
The addition of sulphuric acid to alkenes is also regioselective as it takes place according to Markovnikov’s rule. For example, propene reacts with sulphuric acid to form mainly isopropyl hydrogen sulphate rather than n-propyl hydrogen sulphate:
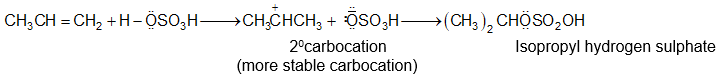
Reaction is simply carried out by bringing the two reactants into contact: a gaseous alkene is bubbled into the acid while a liquid or solid alkene is stirred or shaken with the acid. The result, a clear solution, as alkyl hydrogen sulphates are soluble in sulphuric acid. The alkyl hydrogen sulphates are deliquescent solids, and are difficult to isolate.
Examples
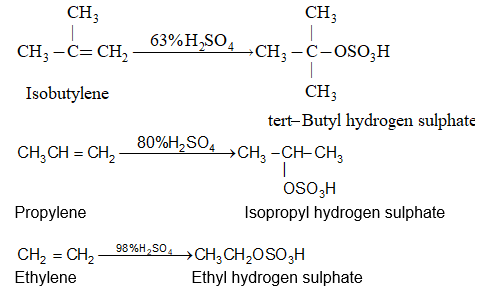
The concentration of sulphuric acid required for reaction depends upon the nature of the alkene involved or to be particular on the nature of carbocation intermediate. Like alkyl sulphonates (RSO3H), these compounds are esters: esters of sulphuric acid, just as alkyl sulphonates are esters of sulphonic acids.
Purification through alkyl hydrogen sulphates
The fact that alkenes dissolve in cold conc. sulphuric acid to form the alkyl hydrogen sulphates is made use of in the purification. For examples, alkanes or alkyl halides, which are insoluble in sulphuric acid, can be freed from alkene impurities by washing with cold conc. sulfuric acid. For this purpose, a gaseous alkane (or alkyl halide) is bubbled through several bottles of sulphuric acid, and a liquid or solid alkane (or alkyl halide) is shaken with sulphuric acid in a separating funnel.
Alcohols from alkyl hydrogen sulphates
An alcohol bearing the same alkyl group as the alkyl hydrogen sulphate can be obtained by diluting the sulphuric acid solution of the alkyl hydrogen sulphate with water, followed by heating. The ester is cleaved by water to form the alcohol and sulphuric acid, and is said to be hydrolyzed. The overall result of the addition of sulphuric acid to an alkene followed by hydrolysis is the Markovnikov’s addition of H- and -OH:
This sequence of reactions is an excellent method for the large – scale manufacture of alcohols, since alkenes are readily obtained by the cracking of petroleum.
Illustration 14: In one industrial synthesis of ethanol, ethene is first dissolved in 95% sulphuric acid. In a second step water is added and the mixture is heated. Outline the reactions involved.
Solution:
Illustration 15: Alkyl hydrogen sulphates can be easily hydrolyzed to alcohols by heating them with water. Which of the following alcohols can be made by this method?
(I) Isopropyl alcohol
(II) n–Propylalcohol
(III) Isobutyl alcohol
(IV) Tert-butyl alcohol
a) I, III b) II, III c) I, IV d) II, IV
Solution: The addition of sulphuric acid follows Markovnikov’s rule consequently, certain alcohols can’t be obtained by this method. Isopropyl alcohol can be made, but not n-propyl alcohol tert-butyl alcohol, but not isobutyl alcohol. Thus, Ans. is (C)
5) Addition of water (Hydration of alkene):
We have just seen that alkenes may be hydrated to alcohols by absorption in concentrated sulphuric acid followed by hydrolysis of the alkyl hydrogen sulphate. However, alkenes may also be catalytically hydrated (directly) in dilute acid solution, as water adds to (the more reactive) alkenes in the presence of acids to yield alcohols:
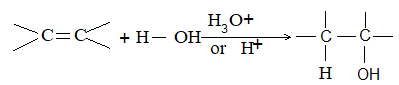
The acid-catalyzed addition of water to the double bond of an alkene (called direct hydration of an alkene) is a method for the preparation of low-molecular-weight alcohols. The acids most commonly used to catalyze the hydration of alkenes are dilute aqueous solutions of sulphuric acid and phosphoric acid.
The acid-catalyzed hydration of alkene is regioselective as the addition of water to the double bond follows Markovnikov’s rule.
Examples:
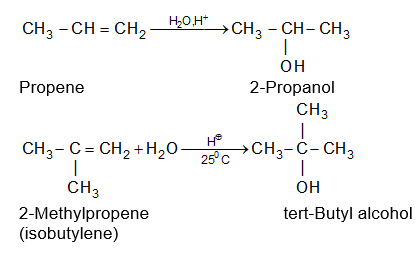
Since these reactions follow Markovnikov’s rule, acid catalyzed hydrations of alkenes do not yield primary alcohols except (in the special case of) the hydration of ethene:
Mechanism for the acid-catalyzed hydration of alkenes:
The mechanism for the acid-catalyzed hydration of an alkene is just the reverse of the mechanism for the hydration of an alcohol.
Step 1: The alkene donates p electron pair to a proton to form the more stable carboaction:
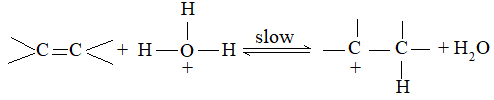
Step 2: The carbocation reacts with a molecule of water to form a protonated alcohol:

Step 3: A transfer of a proton (from protonated alcohol) to a molecule of water leads to the product:
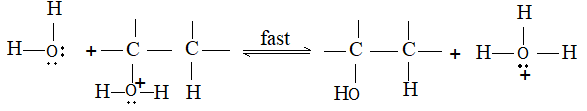
6) Oxymercuration–demercuration: Hydration of alkenes, directly or via the alkyl hydrogen sulphates, proceeds through the intermediate carbocations. If these are capable of undergoing rearrangement ‘unexpected’ alcohols may be obtained from the alkene.
A useful laboratory method for synthesizing alcohols from alkenes that prohibits (avoids) rearrangement is two-step method called oxymercuration–demercuration.
Alkenes react with mercuric acetate in the presence of tetrahydrofuran (THF) and water to yield hydroxy alkyl mercurial compounds which on reduction in aqueous sodium hydroxide with sodium borohydride produce alcohols.
Step I Oxymercuration
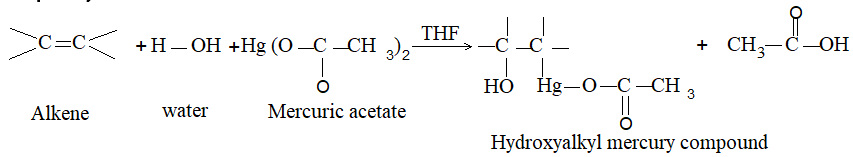
Step II Demercuration
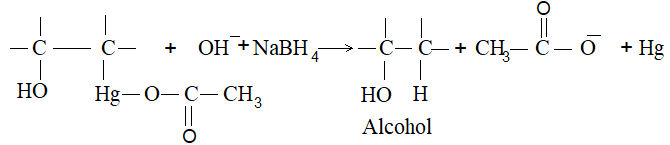
Oxymercuration-demercuration is highly regioselective. The overall result, the addition of the elements of water, H – and -OH, is in accordance with Markovnikov’s rule. The H- gets attached to the carbon atom of the double bond with the greater number of hydrogen atoms:
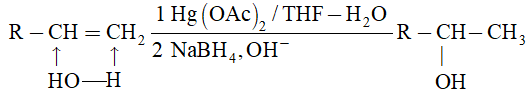
The acetate group, is often abbreviated -OAc, infact the acetyl group, , as -Ac
Examples
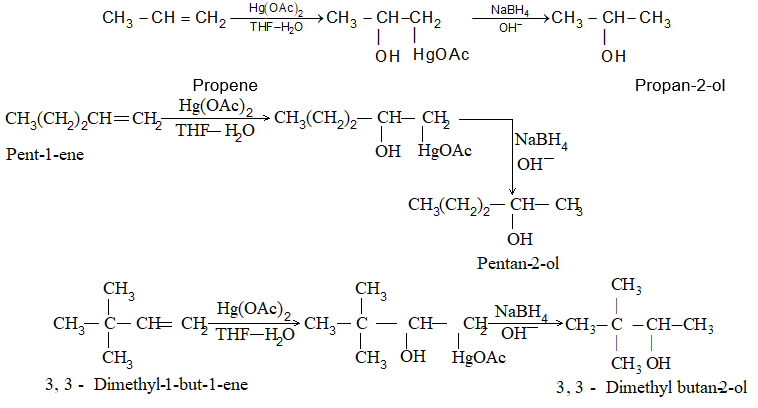
7) Hydroboration–Oxidation: Indirect hydration of an alkene in an anti-Markovnikov sense can be achieved through a two-step method called hydroboration–oxidation.
Alkenes react rapidly with diborane (B2H6) in ether or a solution of borane in tetrahydrofuran (BH3:THF) at room temperature to yield trialkylboranes (R3B), which on oxidation and hydrolysis produce alcohols.
Step I : Hydroboration
It involves addition of BH3 (or, in following stages, BH2R and BHR2) to the double bond, with hydrogen getting attached to one doubly bonded carbon, and boron to the other:
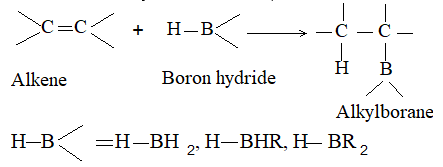
Step II: Oxidation
It involves oxidation and hydrolysis of the alkyl borane intermediate to an alcohol and boric acid:
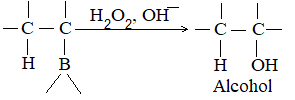
Thus, the two-stage reaction method of hydroboration – oxidation results (overall) in the addition of the elements of water, H- and -OH, to the carbon – carbon double bond of alkene. As a result, the alkene is converted into alcohol.
Orientation of hydroboration – oxidation:
Hydroboration-oxidation is highly regioselective. However, the alcohol formed here is exactly opposite to the one formed by direct/indirect hydration or by oxymercuration – demercuration. The anti-Markovnikov regiochemistry of the hydroboration – oxidation is illustrated by the following examples:
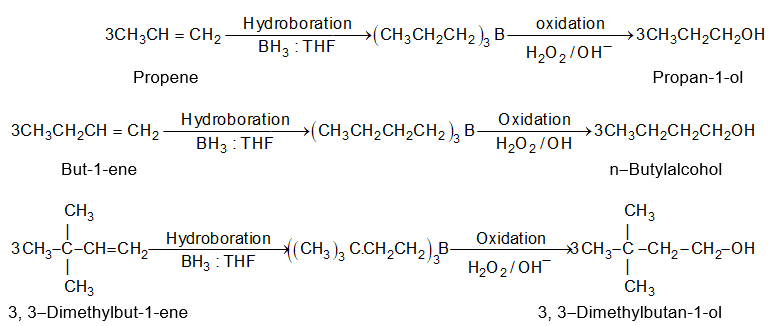
Thus, the hydroboration-oxidation process yields alcohols corresponding to anti-Markovnikov addition of water to the carbon-carbon double bond.
The following example nicely illustrates the important fact that Hydroboration – Oxidation takes place with syn stereochemistry as well as follows anti-Markovnikov regiochemistry:
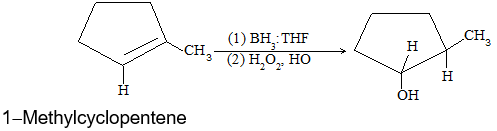
8) Halohydrin formation: If the halogenation (addition of chlorine or bromine) of an alkene is carried out in aqueous solution (i.e., in the presence of water) rather than in carbon tetrachloride, a nonnucleophilic solvent, the chief product of the overall reaction is not a vic-dihalide; instead it is a compound containing halogen and hydroxyl on adjacent carbon atoms. These compounds are thus haloalcohols (chloroalcohols or bromoalcohols) commonly referred to as halohydrins (chlorohydrins or bromohydrins):
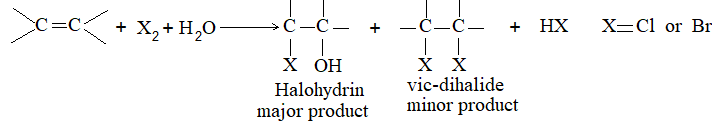
In this case, the molecule of the solvent (H2O) has become the reactant, too.
Examples

The reaction is highly regioselective. With unsymmetrical alkenes, the hydroxyl group adds on to the carbon atom joined to the least number of hydrogen atoms.
Aqueous solutions of hypohalous acid also add on to alkenes in the presence of strong acids to form halohydrins. For example
Halohydrins regenerate the alkene on treatment with zinc dust in acetic acid.
9) Addition of nitrosyl halide:
10) Dimerization (Addition of alkenes):
Under suitable conditions, isobutylene is converted by sulphuric or phosphoric acid into a mixture of two alkenes, each containing exactly twice the number of carbon and hydrogen atoms as the original alkene, isobutylene:
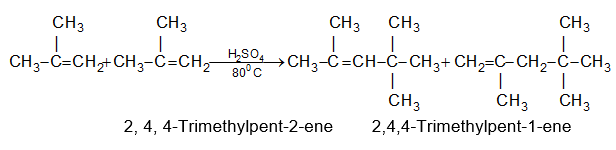
11) Alkylation (addition of alkanes):
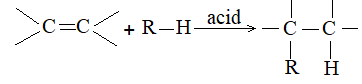
When isobutylene and isobutane are allowed to react in the presence of concentrated sulphuric acid or hydrofluoric acid, they form (directly) 2, 2, 4-trimethylpentane:
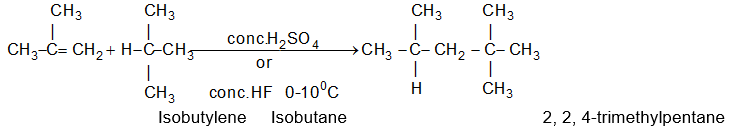
The net result of this reaction is addition of an alkane to an alkene.
12) Oxidation of alkenes:
a) Hydroxylation (Formation of 1, 2–diols) Alkenes are readily hydroxylated (i.e., add on hydroxyl groups), by certain oxidizing agents, to form 1, 2-diols: dihydroxy alcohols carrying the two -OH groups on adjacent carbons. They are also called sometimes as glycols. The hydroxylation reaction results in the addition of two hydroxyl groups to the double bond:

Hydroxylation may be effected
i) By means of cold dilute alkaline (1%) aqueous solution of potassium permanganate [Baeyer’s reagent]. The result is syn (or cis) hydroxylation. For example
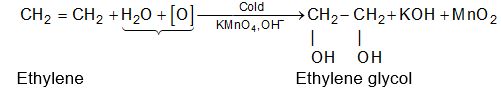
Decolorisation of KMnO4 solution is used as a test for unsaturation. (Baeyer test).
The mechanism of hydroxylation with permanganate proceeds via a cyclic intermediate. This results in syn addition of the oxygen atom.
ii) By means of osmium tetroxide. This compound is widely used to synthesize 1, 2-diols (glycols). It adds very readily to the carbon-carbon double bond of an alkene at room temperature. The mechanism involves cyclic intermediate that results in syn addition of the oxygen atoms:
These cyclic compounds, called osmic esters, on refluxing with aqueous ethanolic sodium hydrogen sulphite or sodium sulphite are hydrolysed to 1, 2-glycols (cis-glycols)
Cleavage at the oxygen-metal bonds takes place without altering the stereochemistry of the two new C-O bonds.
The syn stereochemistry of these dihydroxylations is clearly apparent in the reaction of cyclopentene:

b) Oxidative cleavage
Alkenes can be oxidatively cleaved using potassium permanganate or ozone. Potassium permanganate (KMnO4) is used for strong oxidation while ozone (O3) is used for mild oxidation.
i) Cleavage with hot alkaline potassium permanganate In a cleavage reaction the carbon-carbon double bond is completely broken and the alkene molecule is converted into smaller molecules.
Treatment of an alkene with hot alkaline potassium permanganate results in the cleavage of the double bond. The nature of the products depends upon the structure of alkene:
Unsubstituted alkene carbons are oxidized to carbon dioxide and their hydrogens into water:
Alkenes with monosubstituted carbon atoms are oxidatively cleaved to salts of carboxylic acids:
Disubstituted alkene carbons are oxidatively cleaved to ketones:
The following examples illustrate the above results:
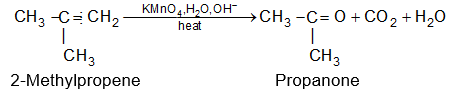
Cleavage occurs via intermediate formation of a 1, 2-diol. Undesired cleavage of this type limits the use of KMnO4 as a reagent for 1, 2-diol synthesis.
Besides being used for desired oxidative cleavage, KMnO4, can be employed in the chemical test for detecting the presence of unsaturation in an unknown compound. Solution of KMnO4 is purple; if an alkene (or even an alkyne) is present the purple color is discharged and a brown ppt. of manganese dioxide (MnO2) forms during oxidation.
The oxidative cleavage of alkenes can be used to establish the location of the double bond in an alkene chain or ring. For this purpose, we must work backward from the products to the reactant that leads to those products.
Illustration16: An unknown alkene of molecular formula C8H16, on oxidation with hot alkaline KMnO4 yielded propanoic acid and pentanoic acid. Find the structure of the alkene. Solution:
Join the carboxylic acid carbons by a double bond to get the alkene. Thus, the unknown alkene must have been cis- or trans-oct-3-ene and oxidative cleavage must have taken place as follows:
(Either cis -or trans-3-octene)
Illustration 17: An unknown alkene with the formula C7H12 on oxidation with hot alkaline KMnO4 yielded only the following product:
What is the structure of this alkene?
Solution:
Since the oxidative cleavage leads to just one product which contains the same number of carbon atoms as the reactant, the unknown alkene must have a double bond contained in a ring. Oxidative cleavage of the double bond results in the opening of the ring. To create the ring, join the carbonyl acid carbons by a double bond.
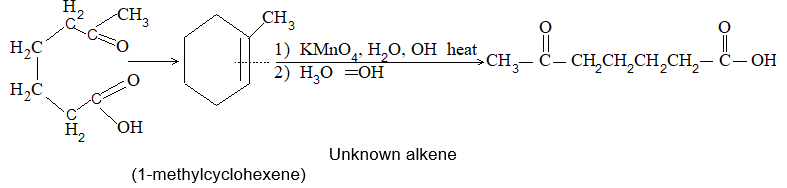
ii) Oxidative cleavage by Ozonolysis
The classical reagent for cleaving the carbon-carbon double bond is ozone. Treatment of an alkene with ozone followed by workup with zinc and acetic acid (or dimethyl sulfide, (CH3)2S) results in oxidative cleavage. This process is called ozonolysis (cleavage by ozone). It occurs in two stages: Stage I; Addition of ozone to the double bond to form an ozonide. For this purpose, the ozone gas is bubbled into a solution of the alkene dissolved in a solvent that is unaffected by ozone (e.g. chloroform, carbon tetrachloride, glacial acetic acid, light petrol). Evaporation of the solvent leaves the ozonide as a viscous oil.
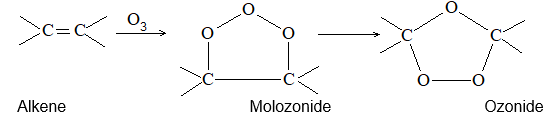
Stage II Reductive hydrolysis of the ozonide to yield the cleavage products. Ozonides are very unstable compounds, and low-molecular-mass ozonides often explode violently in the free state. Because of this behavior, they are not usually isolated (or purified) but are reduced directly (to the carbonyl compounds) by treatment with zinc and acetic acid (HOAc) or dimethyl sulphide:
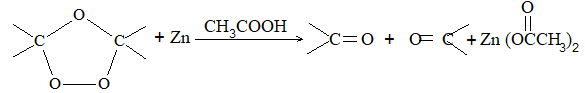
In the cleavage products a doubly bonded oxygen gets attached to each of the originally doubly bonded carbons.
Ozonolysis results in milder oxidative cleavage of alkenes than potassium permanganate. Thus, unsubstituted alkene carbons are oxidized to formaldehyde, rather than to carbon dioxide as with KMnO4. Similarly monosubstituted alkene carbons are oxidized to aldehydes, higher rather than to carboxylate salts as with alkaline KMnO4. However, disubstituted alkene carbons are oxidized to ketones, the same as with KMnO4.
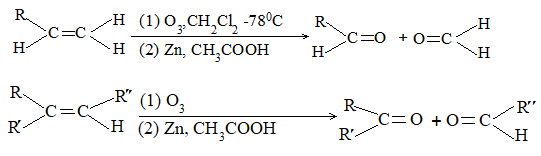
A -H atom attached to the double bond is not oxidized to -OH as it was with permanganate oxidation.
To form ozonolysis products remove the carbon -carbon double bond and attach oxygen to each of the originally bonded carbons:
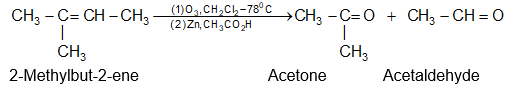
Besides being used to synthesize carbonyl compounds, ozonolysis can be used to determine the location of double bonds in alkenes through structural analysis of the cleavage products.
To create the structure of the original alkene just write the cleavage products, the carbonyl compounds, with their oxygen’s facing towards each other. Now remove the oxygen’s and join the carbonyl carbons by a double bond.
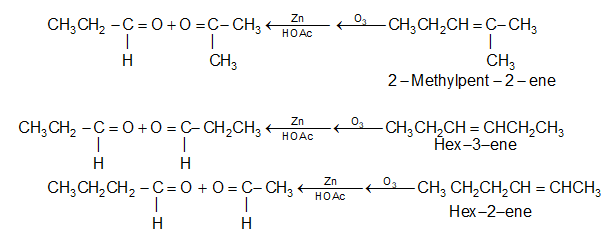
13) Polymerisation: Under different conditions, alkenes polymerise to form many important materials used in our daily life. Everybody is familiar with polyethene bags and polyethene sheets. When ethylene is heated under pressure with oxygen, a compound of high molecular mass (about 20,000) is obtained:
It is essentially an alkane with a very long chain. Since this compound is made up of many ethylene units, it is aptly called polyethylene (or polyethene). It is familiar to almost everyone as the plastic material of packaging films.
The formation of polyethylene is a simple example of the process called polymerization : the joining together of many small molecules to form very large molecules. The compound made up of these very large molecules is called a polymer (Greek: poly + meros, many parts). The simple compounds from which polymers are made are called monomers (mono, one). Other alkenes also undergo polymerization:
Polymers are extensively used for the manufacturing of plastic bags, squeeze bottles, refrigerator dishes, toys, pipes, radio and T.V. cabinets etc. Polypropene is used for the manufacture of milk crates, plastic buckets and other moulded articles.
14) Halogenation (Allylic substitution):
Instead of addition reactions with the halogens, the alkenes may undergo substitution provided the right conditions are used.
When propene reacts with chlorine or bromine at low temperatures, the reaction that takes place is the usual addition of halogen to the double bond:
Addition reaction
However, when propene reacts with chlorine or bromine at very high temperatures or under conditions in which the concentration of the halogen is very small, the reaction that occurs is a substitution.
Substitution reaction
In this substitution a halogen atom replaces one of the hydrogen atoms of the methyl group of propene. These hydrogen atoms are called the allylic hydrogen atoms, and the substitution reaction is known as an allylic substitution.
Allylic hydrogen atom and allylic substitution are general forms.
Allylic chlorination: At a temperature of 400-6000C, a mixture of gaseous propylene and chlorine yields mainly the allylic substitution product, 3-chloroprop-1-ene, known as allyl chloride:
Allylic Bromination Propene undergoes allylic bromination on treatment with N-bromosuccinimide (NBS) in CCl4 in the presence of light or peroxides:
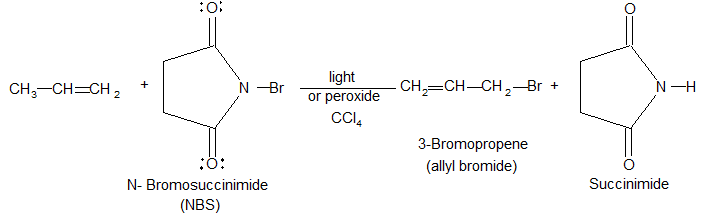
NBS functions simply by providing a constant, low concentration of bromine.
Methods of Preparation of ethylene (or ethene):
Ethylene may be prepared by most of the general methods of preparation.
1) Controlled hydrogenation of acetylene: Acetylene on partial reduction with calculated amount of dihydrogen gas in the presence of Lindlar’s catalyst (Pd-BaSO4 partially poisoned with quinoline) yields ethylene:
2) Dehalogenation of ethylene bromide: When ethylene bromide (or 1, 2-dibromoethane) is heated with zinc dust and methanol, it undergoes dehalogenation (i.e. the removal of the two bromine atoms) to yield ethylene.
3) Dehydrohalogenation of ethyl halides: When ethyl halide (chloride, bromide or iodide) is heated with alcoholic potash (Potassium hydroxide dissolved in alcohol say, ethanol), it undergoes dehydrohalogenation (i.e. the removal of the elements of hydrogen halide, H- and -X, from the adjacent carbon atoms) to yield ethylene.
4) Dehydration of ethyl alcohol: When passed over heated alumina at 3500C, ethyl alcohol undergoes dehydration (i.e. the elimination of the elements of water, H- and -OH, from the adjacent carbon atoms) to produce ethylene:
Ethyl alcohol is also dehydrated, by the action of concentrated sulphuric acid at 160-1700C, to yield ethylene:
5) Industrial method: Ethylene is prepared industrially by heating ethane (obtained from natural gas):
Physical properties:
1) At room temperature, ethene is a colourless, odourless, non-poisonous gas.
2) Melting of solid ethene and boiling of liquid ethene occur at very low temperatures: m.p. -1690C, p. -1020C.
3) In agreement with the rule of thumb that “like dissolves like” it is sparingly soluble in water but readily soluble in organic solvents such as benzene and ether.
Chemical properties: Ethene mainly undergoes electrophilic addition reactions where the pi bond of the carbon-carbon double bond is broken and two strong sigma bonds are formed in its place:
1) Addition of dihydrogen (catalytic hydrogenation): Ethene is readily hydrogenated under pressure in the presence of a catalyst. Finely divided platinum and palladium are effective at room temperature; nickel on a support (Sabatier–Senders reduction) requires a temperature between 2000C and 3000C; Raney nickel is effective at room temperature and atmospheric pressure:
2) Addition of halogens: Ethene reacts rapidly with chlorine and bromine in nonnucleophilic solvents to form vicinal dihalides:
X = Cl or Br
3) Addition of hydrogen halides: Ethene adds up hydrogen halides (HCl, HBr and HI) to form the corresponding ethyl halides:
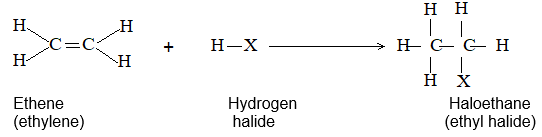
This addition reaction is carried out by dissolving both the hydrogen halide and ethene in a solvent such as acetic acid.
4) Addition of hypochlorous acid: Hypochlorus acid adds on ethene to form ethylene chlorohydrin:
Usually the reaction is carried out by treating the ethene with chlorine-water.
5) Oxidation reactions: Ethene undergoes a number of reactions in which the carbon – carbon double bond is easily oxidized. The following are the two important oxidation reactions of ethene:
a) Dihydroxylation Ethylene reacts with cold dilute alkaline (1%) aqueous solution of potassium permanganate to yield ethylene glycol:
This reaction serves as a chemical test for the presence of unsaturation in an unknown compound (Baeyer’s test). Solution of potassium permanganate is purple. If an alkene (or an alkyne) is present the purple color is discharged and a brown precipitate of manganese dioxide (MnO2) forms as the oxidation takes place.
b) Epoxidation Ethylene gets oxidized to ethylene oxide (epoxide) by air (or oxygen) at a temperature of 200-4000C in the presence of silver catalyst:
6) Ozonolysis Ethylene adds on ozone to give a cyclic compound called ozonide which on hydrolysis (with water) in the presence of zinc dust gives formaldehyde:
Ozonolysis is used to determine the location of double bonds in unsaturated compounds.
7) Combustion Ethylene readily burns on heating in excess of air or oxygen and gets completely oxidized to carbon dioxide and water with the evolution of heat:
8) Formation of mustard gas By the action of sulphur monochloride, ethylene is converted into mustard gas [bis (2-chloroethyl) sulphide, 2, 2¢ -dichlorodiethyl sulphide, b.p. 2150C]:
Mustard gas is a poison and a vesicant.
a) Polymerisation The process of joining together of many small molecules to form very large molecules with or without elimination of small molecules such as water, is called polymerization. The compound composed of these very large no. of molecules is called a polymer (Greek : Poly + meros, many parts). Plastic materials which we use in our daily life are made through polymerization. When ethylene is heated (at 2000C) under pressure (1500-2000 atm.) with oxygen, polyethene (the plastic material of packaging films) is obtained:
or Polymerization of substituted ethylenes, H2C = CH – X (X = Cl, CN, OCOCH3) yields compounds which are used in many ways.
Uses of ethylene
Ethylene is used
1) in the preparation of mustard gas
2) to make ethylene glycol which is used as an antifreeze
3) in the manufacture of ethyl alcohol, polythene etc.
Exercise 4: (i) The alkene which on ozonolysis yields only acetone is
(A)
(B)
(C)
(D)
(ii) Of the following which will have a zero dipole moment?
(A) 1, 1-dichloroehtyl lene
(B) cis-1, 2-dichloroethylene
(C) trans-1, 2-dichloroethylene
(D) None of these
(iii) Ethylene reacts with air under pressure in presence of silver catalyst at 2500C to form
(A) thylene glycol
(B) formaldehyde
(C) acetaldehyde
(D) epoxide
ALKYNES
Alkynes or acetylenes are unsaturated hydrocarbons whose molecules contain one carbon-carbon triple bond. The common name for this homologous series is acetylenes, after the first member, CH º CH, acetylene. They have the general formula CnH2n–2 and the triple bond, the functional group of the family, is known as the ‘acetylenic bond’.
Like the double bond, it is unsaturated and highly reactive towards the reagents that attack the double bond, and towards some others besides. Many alkynes have been found in nature.
Structure of acetylene and the carbon–carbon triple bond
The simplest and stable member of the alkyne family is ethyne, C2H2, which is popularly known as acetylene. Completion of octet by every atom is possible in a structure in which the carbon atoms share 3 pairs of electrons, and hence are joined by a triple bond:
Acetylene Thus, the carbon-carbon triple bond is the distinguishing feature of the alkyne structure.
Quantum mechanical picture
To form bond with two other atoms, each carbon atom of ethyne makes use of two identical sp hybrid orbitals, formed by the mixing of one s and one p orbital:
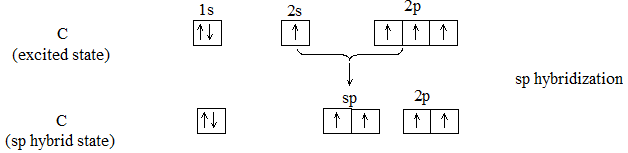
These sp orbitals lie along a straight line that passes through the carbon nucleus. This linear arrangement permits the hybrid orbitals to be as far apart as possible (VSEPR theory). Thus the angle between the two orbitals is 1800, and two linear bonds are formed through these orbitals. When the two carbons and the two hydrogens of acetylene arrange themselves to permit maximum overlap of orbitals, the following structure is obtained (only bonds are shown):
Both carbon-hydrogen and carbon-carbon bonds are cylindrically symmetrical about a line joining the nuclei, and are therefore s bonds. Carbon-carbon sigma () bond is formed by the head-on overlapping of the two sp hybridized orbitals of the two carbon atoms. The remaining sp hybridized orbital of each carbon atom overlaps along the internuclear axis with the 1s orbital of each of the two hydrogen atoms to form two C-H sigma bonds. The H-C-C (or C-C-H) bond angle is 1800. The molecule is still incomplete. To form the sp hybrid orbitals, each carbon atom has used only one of its three p orbitals and thus has two unused pure p orbitals. Each of these consists of two equal lobes, whose axis lies at right angles to the axis of the other p orbital as well as to the line of the sp hybrid orbitals. Each p orbital is occupied by a single electron.
The sum of two perpendicular p orbitals is not four spherical lobes, but a single doughnut-shaped cloud.
The 2p orbitals of one carbon atom are parallel to the 2p orbitals of the other carbon atom. Thus they undergo lateral (or sideways) overlap to permit sharing (or pairing) of electrons. Two pi () bonds are formed, which together make a single cylindrical sheath about the line joining the nuclei.
Thus ethyne molecule consists of one C-C bond, two C-H bonds and two C-C bonds. The carbon-carbon “triple bond” is made up of one strong s bond and two weaker p bonds. It has a total strength of 198 kcal (or 827.64 kJ mol–1). It is stronger than the carbon-carbon double bond of ethylene (163 kcal or 610.7 kJ mol–1) or the carbon-carbon single bond of ethane (88 kcal or 341.1kJ mol–1), and therefore shorter than either.
Verification of the quantum mechanical structure
Electron diffraction, X-ray diffraction, and spectroscopy show acetylene to be a linear molecule. The CºC distance (or the carbon-carbon triple bond length) in acetylene is 1.21 Å, less when compared with 1.34Å, the C=C distance (or the carbon-carbon double bond length) in ethylene and 1.53Å, the C-C distance (or the carbon-carbon single bond length) in ethane.
The linearity of the triple bond does not allow geometrical isomerism at all!
The C-H distance in acetylene is 1.08 Å, shorter than the C-H distance in ethylene, 1.103Å. The explanation lies in the nature of orbitals used for bonding. Because of their greater s character, sp orbitals are smaller than sp2 orbitals and hence sp-hybridized carbon of acetylene forms shorter bonds than sp2-hybridized carbon of ethylene.
Nomenclature
In common system, alkynes are named as derivatives of acetylene. In IUPAC system, they are named as derivatives of the corresponding alkanes replacing ‘ane’ by the suffix ‘yne’. The position of the triple bond is indicated by the first-triply bonded carbon.
Illustration 18: Write the structures and IUPAC names of the following compounds:
a) Methylacetylene b) Ethylacetylene c) Dimethylacetylene
Solution:
a) , Propyne
b) , But-1-yne
c) , But-2-yne
Isomerism
Alkynes exhibit structural isomerism. Ethyne and propyne have got only one structure but for butyne two structures are possible:
They are known as position isomers as they differ in their structures due to the position of the triple bond.
Illustration19:
In how many ways the structure for the alkyne with molecular formula C5H8 can be constructed?
Solution:
First arrange five carbon atoms with a continuous chain and then with a side chain.
I) Pent-1-yne
II) Pent-2-yne
III) 3-Methylbut-1-yne
Structures I and II are position isomers while structures I and III or II and III are chain isomers.
Illustration20: Write structures and IUPAC names of all possible structurally isomeric alkynes corresponding to C6H10. What type of structural isomerism is exhibited by different pairs of isomers?
Solution: First, explore all possibilities for the straight-chain skeleton:
a)
b)
c)
Now explore all possibilities for the branched-chain skeleton with one side chain.
d)
e)
f)
Next explore all possibilities for the branched chain skeleton with two side chains: Different pairs exhibit position and chain isomerism.
g)
Acetylene or ethyne, the most important member of this series may be prepared by any of the following methods:
1) Hydrolysis of calcium carbide On industrial scale, ethyne is prepared by the action of water on calcium carbide:
Calcium carbide is prepared by heating quick lime with coke:
Quick lime can be obtained by heating limestone
2) Partial oxidation of methane It is also made industrially by the controlled, high-temperature partial oxidation of methane:
Other industrial methods are:
a) The cracking of methane-ethane mixture
b) Heating a mixture of ethane (or propane) and steam to 1000-13000
3) Dehydrohalogenation Ethylidene dibromide and ethylene dibromide on treatment with alcoholic potassium hydroxide undergo dehydrohalogenation. The reaction proceeds in two steps:
Similarly
Under suitable conditions the intermediate product vinyl bromide may be isolated.
Sodamide in liquid ammonia can be used instead of ethanolic potassium hydroxide.
The yields are usually better since there is less tendency to form by-products.
4) Intramoleculer dehalogenation 1, 1, 2, 2-tetrahaloethane undergo loss of halogen on heating with zinc dust and methanol to yield ethyne:
5) Intermolecular dehalogenation By heating a haloform with silver powder, e.g., iodoform:
6) Kolbe’s electrolytic method Electrolysis of a concentrated aqueous solution of the sodium (or potassium) salt of maleic or fumaric acid yields acetylene:
Physical properties of Acetylene
1) At room temperature, acetylene is a colourless gas and has an ethereal smell when pure. The garlic smell given off by acetylene is due to the presence of impurities like hydrogen sulphide and phosphine.
2) Melting point of solid acetylene is -820C and boiling point of liquid acetylene is -750
3) It is sparingly soluble in water but readily soluble in organic solvents like acetone, alcohol and ether.
4) When compressed or liquefied acetylene is explosive but it solution under pressure (10 atm.) in acetone, adsorbed on some suitable porous material can be handled with safety.
Chemical properties of acetylene
Due to the presence of a triple bond, acetylene is more unsaturated than ethylene. It forms addition products with two or four univalent atoms or groups, never one or three. When two univalent atoms or groups add on to a triple bond, the linear (or diagonal) arrangement changes into the trigonal and the further addition of two univalent atoms or groups changes the trigonal into the tetrahedral arrangement.
Under suitable conditions it is possible to isolate, the intermediate alkene(s). Acetylene is less reactive than ethylene towards electrophilic reagents. This is not expected in view of the fact that the p-electron density in a triple bond is higher than that in a double bond. A possible explanation for decreased reactivity of a triple bond to electrophiles may be due to fact that the intermediate carbocation from acetylene is less stable than the intermediate corbocation from ethylene.
However, acetylene is found to be more reactive than ethylene towards catalytic hydrogenation as the mechanism of this reaction does not involve electrophiles.
Reactions of Acetylene:
1) Addition of dihydrogen: Acetylene adds on dihydrogen under pressure in the presence of a catalyst, the reaction proceeding in two stages:
Most catalysts affect hydrogenation of a triple bond. By using suitable conditions, it is possible to isolate the intermediate alkene one such method makes use of Lindlar’s catalyst which consists of a Pd – CaCO3 catalyst partially poisoned with lead acetate. A better catalyst is Pd-BaSO4 partially poisoned with quinoline.
2) Addition of halogens: Acetylene adds on gaseous chlorine or bromine in the dark to form acetylene di-and tetra-halides:
Similarly
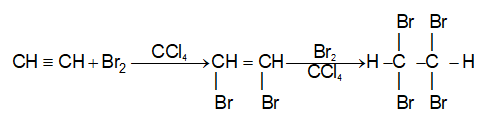
Reddish orange colour of the solution of bromine in carbon tetrachloride is decolorized. This is used as a test for unsaturation. The addition of halogens is catalysed by light and metallic halides. Direct combination of acetylene with chlorine may lead to explosion. This is prevented by the presence of a metal chloride catalyst.
Acetylene reacts with dilute bromine water to produce acetylene dibromide.
With liquid bromine (in the absence of a solvent) acetylene forms acetylene tetrabromide:
Acetylene adds on iodine with difficulty, but if the reaction is performed in ethanolic solution acetylene di-iodide is formed:
The addition of halogens to acetylene is stereoselective as the predominant product is the trans isomer.
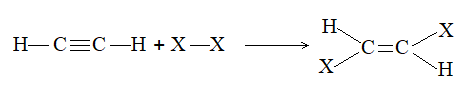
3) Addition of hydrogen halides Acetylene can add on two molecules of hydrogen halides (HCl, HBr and HI) to form gem dihalides, in which the two halogen atoms are attached to the same carbon atom:
Order of reactivity of halogen acids:
HI > HBr > HCl > HF
The reactivity of HF is so low that it adds on only under pressure.
The addition of the halogen acids takes place in the dark but can be catalysed by light or metallic halides. The addition is highly regioselective i.e. in accordance with Markownikov’s rule. For instance, acetylene combines with hydrogen iodide to form first vinyl iodide, and then ethylidene diiodide:
Peroxides have the same effect on the addition of hydrogen bromide to acetylene as they have on alkenes:
4) Addition of water When acetylene is passed into dilute sulphuric acid at 600C in the presence of mercuric sulphate as catalyst, it adds on one molecule of water to form first vinyl alcohol which undergoes tautomerisation to form acetaldehyde as the main product:
5) Addition of hydrogen chloride On treatment with dilute hydrochloric acid at 650C in the presence of mercuric ions as catalyst, acetylene gives vinyl chloride:
6) Addition of hydrogen cyanide In the presence of cuprous chloride in hydrochloride acid as catalyst, acetylene adds on hydrogen cyanide to form vinyl cyanide:
The reaction may also be performed in the presence of cuprous cyanide, Cu2(CN)2, as the catalyst.
Vinyl cyanide, commonly called acrylonitrile, is used in polymer industry, to manufacture Buna-N synthetic rubber, which is a copolymer of vinyl cyanide and butadiene.
Vinyl cyanide is manufactured by passing a mixture of propene, ammonia, steam and air or oxygen over a mixture of oxides of aluminium, cobalt, and molybdenum as catalyst:
7) Addition of acetic acid When passed into warm acetic acid in the presence of mercuric ions as catalyst, acetylene adds on it to form vinyl acetate and ethylidene diacetate:
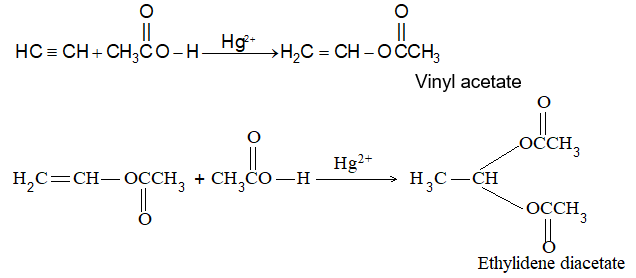
Vinyl acetate Vinyl acetate, a liquid, is used in the plastics industry. Ethylidene diacetate, also a liquid, gives acetic anhydride and acetaldehyde, on heating rapidly to 300-4000C.
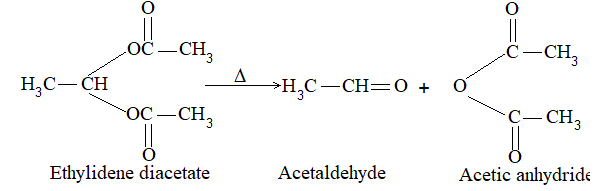
Vinyl acetate is manufactured by
1) passing a mixture of acetylene and acetic acid vapor over zinc acetate on charcoal at about 1700C:
2) by the reaction between ethylene, palladium chloride and sodium acetate in aqueous solution in the presence of cupric chloride:
8) Reaction with nitric acid
Acetylene forms nitroform on reaction with nitric acid in the presence of mercuric ions:
9) Reaction with arsenic trichloride Lewisite is formed when acetylene combines with arsenic trichloride:
10) Vinylation It is the process through which acetylene adds on to compounds containing an active hydrogen atom to form vinyl compounds. For example, when acetylene is passed into methyl alcohol, in the presence of a small amount (1-2 percent) of potassium methoxide, under pressure and at 160-2000C (just high enough to prevent boiling), methyl vinyl ether is formed.
Methyl vinyl ether is used for making the polyvinyl ether plastics.
11) Ethinylation It is the process in which acetylene (or any compound containing the º CH group i.e., a methine hydrogen atom) either adds on a compound containing multiple bonds (unsaturated links) such as a carbonyl compound or reacts with certain hydroxy-compounds through elimination of a molecule of water. For example, acetylene reacts with formaldehyde in the presence of sodium alkoxide as catalyst to form butynediol, together with little amount of propargyl alcohol:
Another example of ethinylation is
12) Addition of ethers Acetylene adds on alkyl bromo – and chloro-methyl ethers, in the presence of the corresponding aluminium halide, to form substituted alkenes:
13) Addition of acyl chloride Acyl chloride adds to acetylene in the presence of the aluminium chloride to form substituted alkene:
14) Addition of hypochlorous acid Acetylene adds on the hypochlorous acid to form dichloroacetaldehyde, when passed into hypochlorous acid solution:
Some amount of dichloroacetic acid, , is formed through the oxidation of dichloroacetaldehyde by the hypochlorous acid.
15) Combustion: Acetylene burns with a luminous smoky flame (due to the high carbon content) on heating in excess of air or oxygen, forming carbon dioxide and water with the evolution of large amount of heat:
Due to luminous nature of the flame, it is used for lighting purposes.
Oxy-acetylene flame (O.A.F), providing a temperature above 30000C, is formed when a mixture of oxygen and acetylene is ignited. OAF blow-pipe is used in welding and cutting process.
16) Ozonolysis
Acetylene adds on ozone to form an ozonide which on hydrolysis in the presence of zinc dust gives glyoxal:
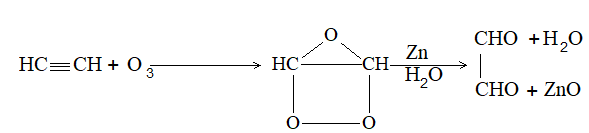
Some amount of formic acid (HCO2H) is also formed.
17) Oxidation Oxidation of acetylene is very much slower than that of ethylene.
18) Polymerisation Acetylene polymerises, to a small extent to benzene, when passed through a heated iron tube:
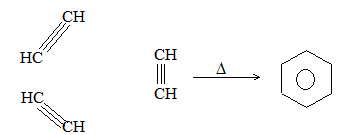
When a mixture of acetylene and ammonia is passed through a red-hot tube, pyrrole is formed, through polymerization:
Pyridine is formed when a mixture of acetylene and hydrogen cyanide is passed through a red-hot tube :
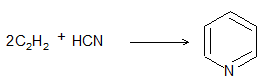
In the presence of Ni (CN)2, acetylene polymerises to cyclooctatetraene:
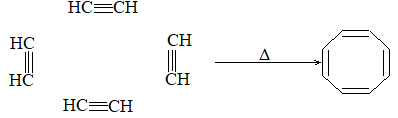
Besides cyclic polymerization, as illustrated above, acetylene undergoes linear polymerization when passed into a solution of cuprous chloride in ammonium chloride to form vinylacetylene and divinylacetylene:
Vinyl acetylene is used to manufacture chloroprene (2-chlorobuta-1, 3-diene). For this purpose, vinylacetylene is passed into concentrated hydrochloric acid in the presence of cuprous chloride and ammonium chloride:
Chloroprene readily polymerises to a rubber -like substance known as neoprene.
19) Acidic nature of acetylene On account of sp, hybridization, acetylenic hydrogens are slightly acidic in nature. Thus, acetylene forms metallic derivatives by replacement of one or both hydrogen atoms. For example, if acetylene is passed over heated sodium, the monosodium as well as disodium acetylides are formed:
The main product is monosodium acetylide if a large excess of acetylene is used. But there are better methods:
i) Passing acetylene into a solution of sodium in liquid ammonia until the blue colour disappers:
ii) Treatment of a fine dispersion of sodium in xylene at 100-1050C with acetylene:
Some interesting properties of acetylides
a) Monosodium derivative absorbs dry carbon dioxide to form the sodium salt of propiolic acid:
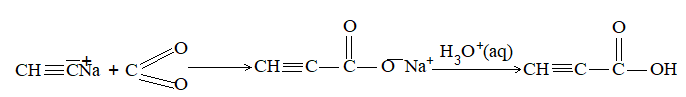
b) Alkali metal acetylide reacts with carbonyl compound to form acetylenic alcohol:
c) An acetylene homologue is obtained when monosodium acetylide is treated with an alkyl halide preferably a bromide:
e.g.
The reaction may be carried further:
For example
Acetylene forms Grignard reagent on reaction with alkylmagnesium halide:
When acetylene is passed into an ammonical solution of cuprous chloride, a red precipitate of cuprous acetylide, Cu2C2 is obtained.
A white precipitate of silver acetylide, Ag2C2, is formed when acetylene is passed into an ammonical solution of silver nitrate:
Both these compounds, when dry, explode when struck or heated.
Physical properties of alkynes
1) Like alkanes and alkenes, alkynes are compounds of low polarity, hence their physical properties are essentially the same as those of the alkanes and alkenes.
2) First three members are gases, the next eight are liquids and the higher ones are solids.
3) All alkynes are colourless and except ethyne, all members are odourless.
4) Being weakly polar in nature, alkynes are almost insoluble in water but quite soluble in the common organic solvents of zero or low polarity; benzene, carbon tetrachloride, ligroin, and ether.
5) They are lighter (i.e. less dense) than water.
6) Their melting and boiling points are very nearly the same as those of alkanes or alkenes with the same carbon skeleton.
7) Their melting points, boiling points and densities show the usual increase with increasing carbon number.
8) The effect of chain-branching are as usual.
Preparation of higher alkynes
Higher alkynes can be made by either of the two techniques:
i) Creating a carbon-carbon triple bond through elimination reactions.
ii) Converting lower members into higher ones i.e. increasing the size of a molecule that already contains a triple bond.
A carbon-carbon triple bond is generated in the same manner as a double bond i.e. eliminating atoms or groups from two adjacent carbons. The reagents used and the atoms or groups eliminated are exactly the same as in the preparation of alkenes.
1) Dehydrohalogenation of dihalogen derivatives of alkanes (alkyl dihalides):
A vicinal dihalide (abbreviated vic-dihalide) is a dihalogen derivative of an alkane carrying the halogens on adjacent carbons (vicinal derives from the Latin vicinus, meaning adjacent). A vicinal dihalide can be synthesized from a suitable alkene by addition of halogen:
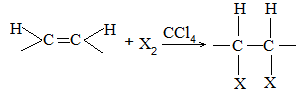
The vicinal dihalide yields an alkyne when subjected to a double dehydrohalogenation reaction:
Thus, alkynes can be synthesized from alkenes via vicinal dihalides.
The dehydrohalogenation occurs in two steps, the first yielding a haloalkene, and the second, the alkyne.
It is a valuable method for preparing unsaturated halides, if carried through only the first stage. These halides, with halogen attached directly to double-bonded carbon, are called vinylic halides. They are very unreactive. Thus, under mild conditions, dehydrohalogenation stops at the vinylic halide stage. More vigorous conditions (i.e., use of a stronger base) are needed for alkyne formation. Depending on the conditions, these two dehydrohalogenations may be carried out as separate reactions.
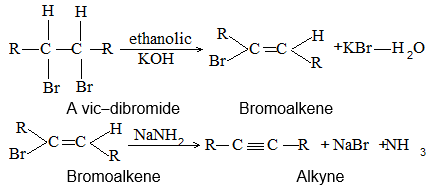
or they may be carried out consecutively in a single mixture:
Sodium amide (or sodamide) is a strong base and thus capable of performing both dehydrohalogenation in a single reaction mixture.
At least two molar equivalents of sodium amide per mole of the dihalide must be used. If the product is a terminal alkyne, then three molar equivalents must be used because the terminal alkyne gets depotonated by sodium amide the moment it is formed in the mixture, consuming the sodamide otherwise needed for the remaining dehydrohalogenation steps.
Dehydrohalogenation with sodamide is usually carried out in liquid ammonia or in an inert medium such as mineral oil.
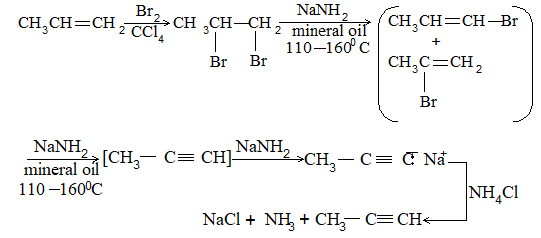
A geminal dihalide (abbreviated gem-dihalide) has two halogen atoms bonded to the same carbon (from the Latin, geminus, twins). A gem-dichloride can be synthesized from a suitable ketone by reaction with phosphorus pentachloride:
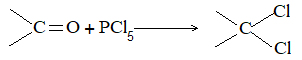
The geminal dihalide also yields an alkyne when subjected to a double dehydrohalogenation reaction:
Example:
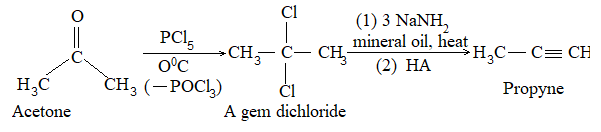
A simple method of introducing a triple bond into an organic compound:
1 butanol but-1-yne
1-Butanol is catalytically dehydrated to but-1-ene, which on treatment with bromine gives 1, 2-dibromobutane, and this, when treated with sodamide in mineral oil gives but-1-yne:
Open-chain 1, 2-dihalides give alkynes rather than conjugated dienes an treatment with strong base like sodamide:
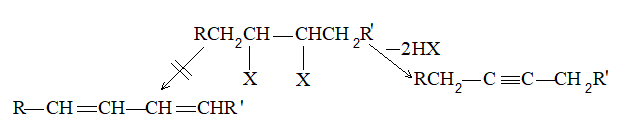
2) Reaction of metal acetylides with primary alkyl halides: We can replace a hydrogen attached to a triply bonded carbon of a terminal alkyne (called an acetylene hydrogen) by an alkyl group. This type of reaction, called an alkylation, is used to convert smaller terminal alkynes into larger alkynes.
The acetylenic hydrogen is weakly acidic and can be removed with a strong base such as sodamide:
Sodium alkynide These reactions almost reach up to completion.
Sodium ethynide and other sodium alkynides can be prepared by treating terminal alkynes with sodamide in liquid ammonia.
Once the acetylenic hydrogen is removed, the alkyne carbon turns into an anion, called an alkynide anion. It can react with a suitable primary alkyl halide, preferably an iodide:
The alkyl group of alkyl halide becomes attached to the triply bonded carbon to generate a new, larger alkyne.
Examples
The alkyl halide used with the sodium alkynide (or the alkynide anion) must be methyl or primary and also unbranched at the beta (second) carbon. Alkynide anions are strong bases, thus, alkyl halides that are secondary or tertiary or are primary with branching at the beta carbon react predominantly by an elimination mechanism to give other products.
Formation of a new carbon-carbon bond by alkylation of an alkynide anion is in itself an important transformation as the alkyne triple bond can be used for further reactions. For instance, hydrogenation of newly synthesized alkyne becomes the formal synthesis of an alkane. For example
3-Methylbut-1-yne It is not appropriate to use propyne and 2-bromopropane for the alkylation step of above synthesis because elimination would predominate as the alkyl halide is secondary.
Reaction of alkynes
The chemistry of alkynes is basically the chemistry of the carbon-carbon triple bond. Due to the availability of the loosely held electrons, alkynes undergo electrophilic addition. Addition of different reagents to alkynes is very much like addition to alkenes, except that here two molecules of reagent can be consumed for each triple bond. By proper selection of reaction conditions, it is usually possible to limit reaction to the first stage of addition i.e. formation of alkenes.
Besides addition, alkynes undergo some reactions that arise due to the acidity of a hydrogen atom held by triply bonded carbon.
1) Addition of hydrogen Depending on the conditions and the catalyst employed, one or two molar equivalents of hydrogen will add to a carbon-carbon triple bond:
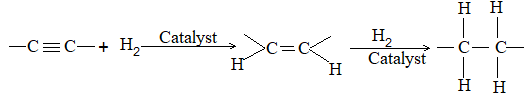
When a platinum catalyst is used, the alkyne usually reacts with two molar equivalents of hydrogen to yield an alkane:
However, hydrogenation of an alkyne to an alkene can be achieved by means of special reagents or catalysts. Unless the triple bond is at the end of a chain, reduction of an alkyne to the double-bond stage can yield either a cis alkene or a trans-alkene. Which of these two isomers predominates depends exclusively upon the choice of reducing agent.
Syn addition of hydrogen leads to synthesis of cis-alkene
An addition that places the parts of the addition reagent on the same side (or face) of the reactant (substrate) is called syn addition.
Almost entirely cis alkene (as high as 98%) is obtained by hydrogenation of non-terminal alkynes (dialkylacetylenes) with specially conditioned catalysts. Metallic palladium deposited on calcium carbonate, conditioned with lead acetate and quinoline called, Lindlar’s catalyst, can be used for syn addition:
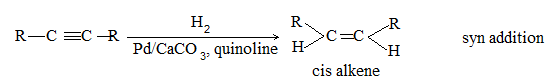
Another heterogeneous catalyst permitting hydrogenation of an internal alkyne to a cis-alkene is the nickel boride compound called P-2 catalyst:
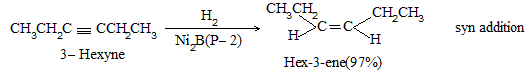
In both the cases, the reaction takes place on the surface of the catalyst, accounting for the syn addition.
The P-2 catalyst can be prepared by the reduction of nickel acetate with sodium borohydride.
Anti addition of hydrogen leads to synthesis of trans alkene
The opposite of a syn addition is an anti addition. An anti addition places the parts of the adding reagent on opposite faces of the reactant.
Predominantly, an anti addition of hydrogen atoms to the triple bond occurs when internal alkynes are reduced with sodium or lithium metal in liquid ammonia or ethylamine at low temperatures:
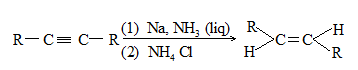
This reaction is called a dissolving metal reduction, as it takes place in solution. The product is mainly trans-alkene.
2) Addition of halogens Alkynes undergo the same kind of addition reactions toward halogens (chlorine and bromine) that alkenes do. However, with alkynes bearing two -bonds the addition may occur once or twice depending on the number of molar equivalents of halogen employed:
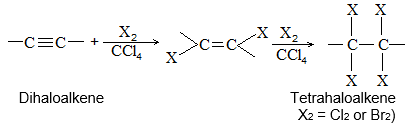
Example
Reddish orange color of the solution of bromine in carbon tetrachloride is decolorized – a test for unsaturation.
By simply adding one molar equivalent of the halogen, it is possible to prepare a dihaloalkene:
The addition of halogens to alkynes is stereoselective:
Additions of chlorine and bromine to alkenes are mostly anti additions and thus trans-dihaloalkenes are formed.
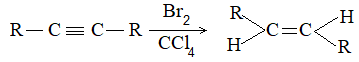
Alkynes are considerably less reactive than alkenes toward the addition of halogens. The lower reactivity of alkynes is due to the greater difficulty in the initial formation of a cyclic halonium ion.
3) Addition of hydrogen halides Depending on whether one or two molar equivalents of the hydrogen halide are used, alkynes react (with hydrogen halides) to form haloalkens or geminal dihalides:
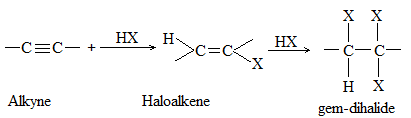
(HX = HCl, HBr, HI)
Order of reactivity: HI > HBr > HCl.
Both stages of additions are regioselective as they follow Markovnikov’s rule. Thus, the hydrogen atom of the hydrogen halide gets attached to that triply bonded carbon atom that has the greater number of hydrogen atoms:
The addition of HBr to an alkyne can also be performed by using acetyl bromide (CH3COBr) and alumina.
Acetyl bromide acts as an HBr precursor by reacting with the alumina to generate HBr. The alumina by its presence increases the rate of reaction.
When peroxides are present in the reaction mixture, anti-Markovnikov addition of hydrogen bromide to alkynes takes place:
Illustration 21: Identify the products ‘X’ and ‘Y’ in the following reaction
Solution:
4) Addition of water (Hydration of alkynes)
Like alkanes and alkenes, alkynes are also immiscible and do not react with water. Like alkenes, alkynes can be hydrated under suitable conditions.
Alkynes add on water to the triple bond readily, provided the reaction is catalyzed by strong acids and mercuric ions (Hg2+). Aqueous solutions of sulphuric acid (42%) and mercuric sulphate (1%) are commonly used for this purpose.
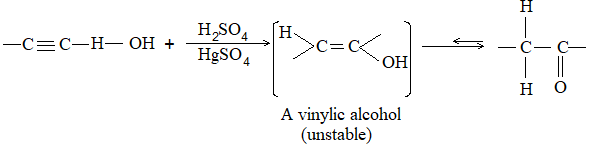
The vinylic alcohol produced initially is generally unstable, it rearranges rapidly to a ketone (or to acetaldehyde, in the case of ethyne). The rearrangement takes place quite easily because of the polarity of the -O-H bond. It involves the loss of a proton from the hydroxyl group, the addition of a proton to the vicinal carbon atom, and the relocation of the double bond.
This kind of rearrangement is acid catalyzed and is known as a tautomerization.
A structure with -OH group attached to a doubly bonded carbon is called an enol (-ene for the carbon-carbon double bond, -ol for alcohol). Vinylic alcohols are typical examples of enols. Whenever one tries to make a compound with enol structure, rearrangement happens and the product is a compound with the carbonyl group. These rearrangemnts are known as keto-enol tautomerizations:
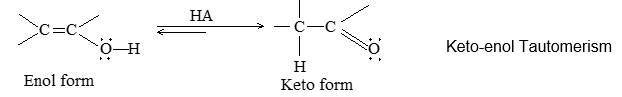
There is an equilibrium between the two forms, but it usually lies very much in favor of the more stable, keto form. Thus, vinylic alcohol is formed initially by hydration of alkyne, but it is rapidly converted into an equilibrium mixture that is almost all carbonyl compound. The addition of water to alkynes is highly regioselective as it follows the Markovnikov’s rule. The hydrogen atom gets attached to the carbon atom with the greater number of hydrogen atoms. When ethyne undergoes addition of water, the main product is an aldehyde:
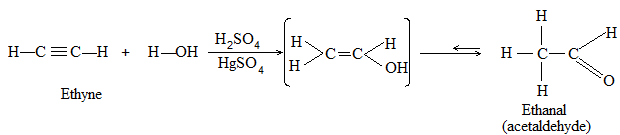
This reaction is often used in the commercial production of ethanal.
When terminal alkynes other than ethyne are hydrated, ketones rather than aldehydes, are the main products: The hydration method is not useful for internal alkynes because a mixture results.
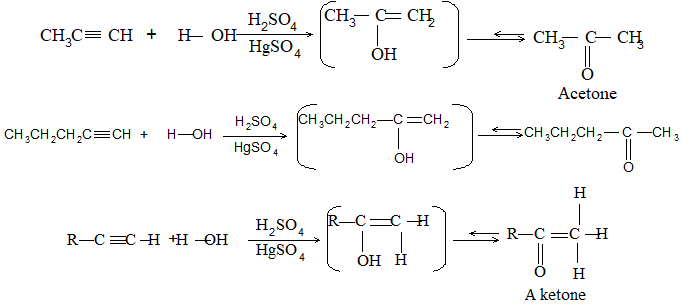
5) Oxidative cleavage of alkynes
a) Like alkenes, alkynes also form ozonides with ozone. These compounds are decomposed by water in the presence of zinc dust to form dicarbonyl compounds:
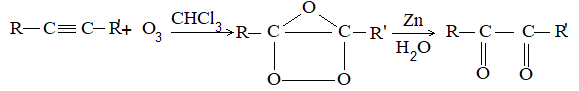
R and R’ may be hydrogens or alkyl groups.
Examples
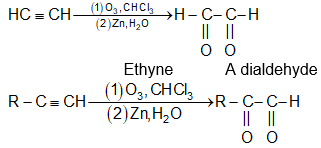
Terminal alkyne
Other than ethyne.
If Zn dust is not used then dicarbonyl compounds are oxidized and cleaved to carboxylic acids by the hydrogen peroxide formed in the reaction:
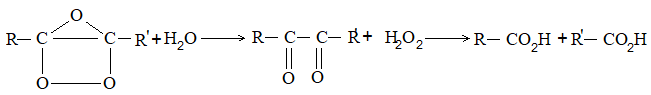
Treatment with alkaline potassium permanganate also leads to cleavage at the carbon-carbon triple bond and the products are carboxylic acids:
6) Acidic character of terminal alkynes (formation of metal acetylides)
Acidity measures the tendency of a compound to lose a proton (hydrogen ion) [Lowry-Brønsted view point]. Considerable acidity is usually shown by compounds in which at least one hydrogen is directly attached to an electronegative atom such as N, O, S and X. The bond holding the hydrogen is considerably polar, and the relatively electron deficient hydrogen can come out as a positive ion.
A triply bonded carbon is more electronegative than a singly or doubly bonded carbon. Consequently hydrogen bonded to triply bonded carbon of acetylene or any terminal alkyne (R – C CH) shows appreciable acidity than those bonded to carbons of an alkene or alkane. The pKa values for ethyne, ethene, and ethane support this point:
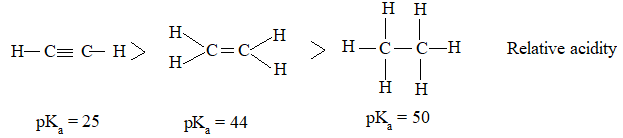
The relative order of basicity of their anions is exactly opposite that of their relative acidity:
Relative basicity
Thus, unlike ethane and ethene, ethyne (acetylene) reacts with sodium metal liberating dihydrogen gas and forming the compound sodium acetylide:
One can also convert ethyne to its conjugate base (a carbanion) by treating it with sodamide in liquid ammonia:
Majority of the alkynes having a proton attached to a triply bonded carbon (called terminal alkynes) have pKa values of about 25. Therefore, they all react with sodamide in liquid ammonia in the same way that ethyne does:
7) Isomerisation Acetylene homologues isomerise under suitable conditions.
a) The triple bond moves towards the centre of the chain when alkynes are heated with ethanolic potassium hydroxide:
2. The triple bond moves towards the end of the chain, when alkynes are heated with sodamide in an inert solvent like paraffin:
This reaction can be used to step up the alkyne series by metal acetylide formation.
Analysis of Alkynes
Alkynes can be analysed by the following methods and observations.
1) They decolourize solution of bromine in carbon tetrachloride without evolution of hydrogen bromide.
2) They decolourize cold alkaline aqueous solution of potassium permanganate.
3) They are not oxidized by chromic anhydride.
4) Amount of unsaturation can be detected by determination of their molecular formula (CnH2n–2).
5) Degree of unsaturation can also be determined by a quantitative hydrogenation : two moles of dihydrogen gas are taken up per mole of a triple bond of the alkyne.
6) Proof of structure is best obtained by degradative method: upon ozonolysis alkynes yield carboxylic acids
Treating alkynes with basic potassium permanganate leads to cleavage at the carbon-carbon triple bond to produce carboxylic acids
7) The following reaction can be used to differentiate terminal alkynes (those with the triple bond at the end of the chain) from non-terminal alkynes: Acidic terminal alkynes form insoluble acetylides on reaction with heavy metal ions, Ag+ and Cu+.
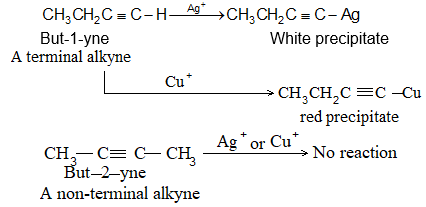
Formation of a precipitate upon treatment of an alkyne with a solution of AgNO3 in alcohol is a clear indication of hydrogen attached to triply bonded carbon. These heavy metal acetylides tend to explode when they are allowed to dry. Thus, they should be destroyed while still wet by warming with nitric acid:
The strong mineral acid displaces the weak acid, alkyne, from its salt.
Under suitable conditions, ethyne can be linearly polymerized to produce polyacetylene (or polyethyne. It is a high molecular weight polyene containing repeating units of and hence can be represented as . Under special conditions, this polymer conducts electricity. This films of polyacetylene can be used as electrodes in batteries. There films are good conductors, lighter and cheaper than the metal conductors.
Illustration 22: Covert acetic acid into benzene.
Solution:
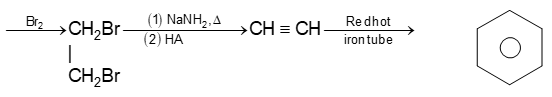
AROMATIC HYDROCARBONS
In the initial development of organic chemistry, chemists found useful to classify all known organic compounds into two broad categories:
a) Aliphatic compounds, and
b) Aromatic compounds
The aliphatic compounds were so named because the first compounds of this class to be studied were the fatty acids (Greek, aliphos, fat). Besides aliphatic compounds, there was a large number of compounds which were obtained from natural sources (essential oils balsams, resins etc.). The structures of these compounds were unknown but they had one thing in common: a pleasant odour. Hence these compounds were (arbitrarily) classified as aromatic (Greek: aroma, fragrant smell). The original meanings of the words “aliphatic” (fatty) and “aromatic” (fragrant) have lost the significance.
The term aliphatic is now applied to any compound that has an open-chain structure and also to any cyclic compound that resembles an open-chain compound. Careful analysis of aromatic compounds indicated that they carried a higher percentage carbon content than the corresponding aliphatic hydrocarbons. It also showed that most of the simple aromatic compounds contained at least six carbon atoms. Further examination showed that when complex aromatic compounds were subjected to various degradive methods, they almost always produced benzene or a derivative of benzene. Whenever, attempts were made to degrade aromatic compounds into compounds with less carbon atoms than six (as in benzene), the entire molecule usually disrupted. It clearly indicated that aromatic compounds were related to benzene. Thus, aromatic compounds are now taken as benzene and compounds that resemble benzene in chemical behavior.
Kekulé was the first to recognize that early aromatic compounds all contain a six-carbon unit and that they retain this six-carbon unit through most chemical transformations and degradations.
Aromatic benzene molecule is a ring of a very special kind. For instance, benzene ring is highly unsaturated but in a majority of reactions, the unsaturation of benzene ring is retained. There are certain ring compounds which differ from benzene in structure but behave very much like benzene. These compounds actually resemble benzene in basic electronic configuration and hence are aromatic, too.
Aromatic compounds containing benzene ring are known as benzenoids and those not containing a benzene ring are known as non-benzenoids. Both are cyclic, but their properties are totally different from those of the alicyclic compounds.
Aromatic hydrocarbons are also known as ‘arenes’.
Aliphatic hydrocarbons (alkanes, alkenes, alkynes, and their cyclic counter parts) undergo mainly free-radical substitution and addition reactions. -Additions reactions take place at multiple bond (double and triple bonds) while free-radical substitution reactions take place at saturated points along the aliphatic chain.
On the other hand, aromatic hydrocarbons are characterized by a tendency to undergo heterolytic (ionic) substitution. These reactions are in fact the characteristic of aromatic rings wherever they appear, irrespective of other functional groups present in the molecule.
The study of aromatic compounds started with the discovery in 1825 of benzene (phene) by the English chemist Michael Faraday. Faraday called this new hydrocarbon “bicarburet of hydrogen”. Mitscherlich (1834) proposed the name benzene. Faraday isolated benzene from cylinders of compressed illuminating gas obtained from the pyrolysis of whale oil.
In 1834, the German Chemist Eilhardt Mitscherlich synthesized benzene by heating benzoic acid with calcium oxide:
Using vapor density measurements, Mitscherlich found that benzene has the molecular formula C6H6. In 1845, benzene was found in coal-tar by Hofmann. This is still a source of benzene and its derivatives.
Commercial preparation of benzene from coal tar
When coal is destructively distilled (heated to a temp. of 1000-14000C in the absence of air in iron retorts), four-fractions are obtained:
1) Coal gas
2) Coal tar
3) Ammonical liquor
4) Coke (solid residue)
Coal tar is a black viscous oily liquid with unpleasant odour. This is heated to remove water and subjected to fractional distillation. The fractions are collected in separate receivers. The number of fractions taken from coal-tar varies. One sample is shown in the following table:
| Name of the fraction | Temperature Range | Density | |
| 1) | Crude light oil or Crude naphtha | Up to 1700C | 0.970 |
| 2) | Middle oil or Carbolic oil | 170–2300C | 1.005 |
| 3) | Heavy oil or Creosate oil | 230 – 2700C | 1.033 |
| 4) | Green oil or Anthracene oil | 270 – 3600C | 1.088 |
| 5) | Pitch (left in retort) | ||
Another sample involves three fractions:
1) Middle oil 2) Heavy oil 3) Pitch
Subsequent treatment depends on the number of fractions taken: The light-oil fraction (first fraction) which is lighter than water is washed with sodium hydroxide (to remove acids such as phenol), then with sulphuric acid (to remove bases) and finally with water. The washed light oil is now subjected to fractional distillation. Various fractions may be taken:
The first fraction collected up to 1100C is known as ’90 per cent benzol. It contains 70 percent benzene, 24 percent toluene and some xylene.
The second fraction collected between 110-1400C gives pure xylene.
The distillate between 140-1700C is known as ‘solvent naphtha or benzine. It consists mainly of xylenes, cumenes, etc. It is used as a solvent for paints, rubber resins etc.
The fraction ‘90 per cent benzol’ gives pure benzene, toluene and xylene on careful fractionation. About a 5% benzene and 8.7 per cent toluene are obtained from the crude light -oil fraction.
Other methods of preparation of benzene
1) Cyclic polymerization of acetylene (Berthelot synthesis) : Passing acetylene gas through red hot iron or copper tube at 873 K:

2) Decarboxylation of aromatic acids Heating sodium salts of acids with sodalime and acids with calcium oxide.
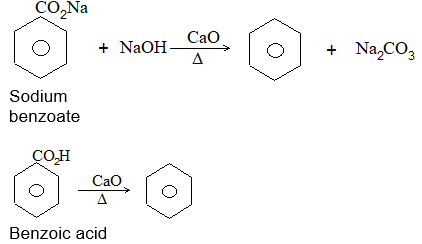
3) Reduction of phenol When distilled with zinc dust, phenol is converted into benzene:

4) From benzene sulphonic acid (desulphonation) Benzene sulphonic acid may be desulphonated to yield benzene, by heating with dilute hydrochloric acid under pressure of 150 – 2000C:
Benzene-d6 (C6D6) is a common solvent, used in NMR studies.
5) From phenyl halides Benzene is formed when phenyl halide undergoes hydrogenolysis with Pd/C/H2, Mg/isopropyl alcohol, and Ni-Al alloy/aq NaOH.

6) From benzenediazonium chloride Replacement of the diazo-group by hydrogen offers a means of preparing benzene. The most reliable method of replacing the diazo-group is by means of hypophosphorous acid. The reaction is catalyzed by cuprous salts.
Another reducing agent available is SnCl2/NaOH:

Exercise 5: (i) Mark the fraction of the coal tar distillation in which benzene and toluene both are present
(A) light-oil
(B) middle – oil
(C) heavy – oil
(D) anthracene – oil
(ii) Which one of the following statements is wrong?
(A) Aromatic compounds are richer in carbon content
(B) Aromatic compounds burn with smoky flame
(C) Aromatic compounds are generally unstable
(D) Aromatic compounds show substitution reactions
(iii) The C – C – C bond angle in benzene is
(A) 900
(B) 600
(C) 1090
(D) 1200
Preparation of benzene and its homologues from petroleum
Aromatic compounds occur naturally in petroleum and can be extracted from it. These days, the main source of aromatic compounds are the non-aromatic constituents of petroleum. This conversion is done by
i) Catalytic reforming or hydroforming:
It is carried out under pressure at 480-5500C in the presence of chromium oxide on an alumina support.
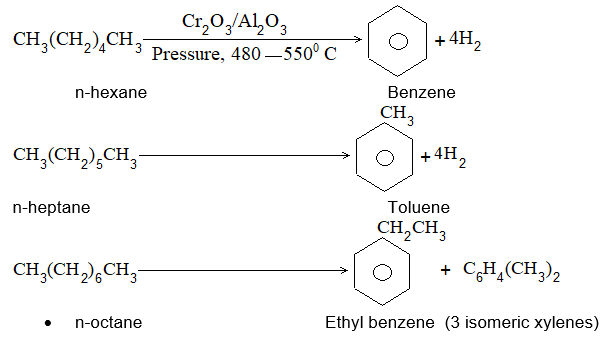
This method is based on three reactions:
1) Dehydrogenation, 2) Cyclisation, and 3) Isomerisation.
The aromatic compounds obtained contain the same number of carbon atoms as the aliphatic starting compounds. The various hydrocarbons are separated by selective solvent process.
Benzene is obtained in far less amount than toluene and the xylenes. Thus, these are converted into benzene by heating with hydrogen under pressure in the presence of a metal oxide catalyst. The process is called hydrodealkylation:
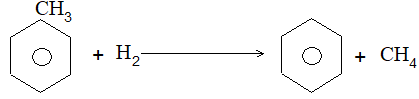
ii) High –temperature cracking in the presence of a catalyst
The following aromatic hydrocarbons could be isolated from the cracking of alkanes: Benzene, toluene, xylenes, naphthalene, anthracene and many other polynuclear hydrocarbons The charging stock is usually cracked at about 650-6800C in tubes packed with metallic dehydrogenation catalysts e.g., chromium oxide on an alumina support.
Physical properties of benzene
1) Benzene is a colourless liquid with a peculiar smell.
2) Melting point of solid benzene is 5.50C, and boiling point of liquid benzene is 800
3) It is inflammable, burns with a smoky (sooty) flame. This property is the characteristic of most aromatic compounds but not of most aliphatic compounds. It is on account of the high carbon content of the aromatic compounds.
4) It is almost insoluble in water but soluble in common organic solvents such as alcohol and ether.
Uses of benzene
1) It is a very good solvent for fats, resins, sulphur, iodine, etc.
2) It is used in dry cleaning.
3) It is also used as a motor fuel (benzol’)
4) It is used for the manufacture of styrene (the monomer used to form polystyrene), adipic acid (used for the synthesis of nylon), phenol, aniline, nitrobenzene, dyes, drugs and insecticides.
Chemical behaviour of benzene
Benzene with the molecular formula of C6H6 (or CnH2n–6) is a highly unsaturated compound. Chemists expected benzene to react accordingly:
a) They expected benzene to react like an alkene by decolorizing bromine in carbon tetrachloride through addition of bromine.
b) They expected that benzene would decolorize cold alkaline aqueous potassium permanganate by being oxidized.
c) They expected that benzene would add hydrogen rapidly in the presence of a metal catalyst.
d) They expected that benzene would add water in the presence of strong acids.
Benzene is a very stable compound. When benzene is treated with bromine in carbon tetrachloride in the dark or with cold alkaline dilute aqueous potassium permanganate or with dilute acids, none of the expected reactions occurs. Benzene does add hydrogen in the presence of finely divided nickel, but only at high temperatures and under high pressures:
Benzene Vs. Cyclohexane
| Reagent | Cyclohexene | Benzene |
| Br2/CCl4 (in the dark, 250C) | Undergoes rapid addition | No addition of bromine |
| KMnO4, 250C | Undergoes rapid oxidation | No oxidation |
| H3O+/H2O, (heat) | Undergoes hydration | No hydration |
| HI | Undergoes rapid addition | No addition |
| H2/Ni | Undergoes rapid hydrogenation at 250C, 20 lb/in2 | Undergoes slow hydrogenation at 100-2000C, 1500 lb/in2 to give cyclohexane |
Under conditions, that cause an alkene to undergo rapid addition, benzene reacts either not at all or very slowly.
The action of halogen (chlorine and bromine) on benzene depends on the prevailing conditions. In bright sunlight, chlorine atoms add on to form benzenehexachloride, C6H6Cl6. In the absence of direct sunlight benzene reacts not by addition but by substitution (benzene substitution).
The reaction is slow but in the presence of a Lewis acid catalyst such as ferric halide, substitution is rapid. Substitution products of benzene are also obtained, when benzene is heated with concentrated sulphuric acid or concentrated nitric acid:
In place of addition reactions, benzene readily undergoes substitution.
Nomenclature: Discussed in phase III
Structure of benzene:
The structure of benzene is built upon the following facts:
1) The molecular formula of benzene is C6H6: Elemental analysis and molecular-weight determination show that the molecular formula of benzene is C6H6.
2) Benzene is a highly unsaturated compound: The number of hydrogen atoms in benzene is eight less than the corresponding alkane. Hence benzene is expected to exhibit unsaturated reactions. It does so in practice:
a) In bright sunlight, benzene adds on chlorine, to form benzenehexachloride, C6H6Cl6, the maximum number of chlorine atoms added is six.
b) Benzene adds on ozone to form a triozonide, C6H6(O3)3.
c) At high temperatures and under pressure, benzene may be catalytically hydrogenated to cyclohexane, C6H12, the maximum number of hydrogen atoms added is six.
3) Unsaturation of benzene is remarkably different from the aliphatic compounds: The following chemical facts clearly show that the pi (p) bonds of benzene behave in a most remarkable fashion in comparison with pi (p) bonds in aliphatic compounds:
d) A solution of chromic acid or alkaline permanganate (both powerful oxidizing agents) has no action on benzene in the cold, but on prolonged heating benzene is broken down into carbon dioxide and water.
e) Halogen acids do not add on to benzene. However, benzene is converted into a mixture of cyclohexane and methylcyclopentane when heated with hydrogen iodide at 2500
f) In the absence of sunlight but preferably in the presence of a halogen carrier ( a Lewis acid catalyst), benzene does react with halogen but most surprisingly it reacts not by addition but by substitution (benzene substitution).
Reactions (a) – (f) lead to the conclusion that benzene contains three pi (p) bonds, but these pi (p) bonds are remarkably different from pi (p) bonds in aliphatic compounds. Probably, this difference leads to ‘aromatic properties’: unusual degree of unsaturation, unusual stability.
The Kekulé structure for benzene
In 1858, August Kekulé, laid the basis of the structural theory – one of the most fundamental theories in chemistry, by proposing two central premises:
1) The atoms in organic compounds can form a fixed number of bonds. The measure of this ability is called valence. Carbon is tetravalent, (i.e., carbon atoms form four bonds), oxygen is divalent while hydrogen and halogens (usually) being monovalent.
2) A carbon atom can use one or more of its valences to form bonds to other carbon atoms. This way, carbon atoms can join to one another to form chains.
In 1865, Kekulé proposed the ring structure for benzene. This structure is still used today but with a different meaning. Kekulé proposed that the carbon atoms of benzene are in a ring, that they are bonded to each other by alternating single and double bonds, and that one hydrogen atom is attached to each carbon atom.
This structure satisfied the requirements of the structural theory that carbon atoms are tetravalent. The structure also contains three pi () bonds as suggested by the reactions (a)-(g).
One can think of other structures consistent with the formula C6H6. Some examples are:
Selection of the most satisfactory of all these should be based upon the evidence of isomer number.
4) For any atom or group of atoms W, benzene yields only one monosubstitution product, C6H5W: When benzene reacts so that one hydrogen atom is replaced by bromine, only one monobromobenzene, C6H5Br, is obtained. That is, only one compound with the formula C6H5Br is found among the products. Similarly, when benzene is chlorinated or nitrated, only one monochlorobenzene C6H5Cl or one nitrobenzene, C6H5NO2 ever results.
This fact puts a severe restriction on the structure of benzene. Two possible explanations can be provided for these observations:
i) Only one of the six hydrogen atoms in benzene is reactive towards these reagents.
ii) All the six hydrogen atoms in benzene are equivalent i.e. each hydrogen must be exactly equivalent to every other hydrogen. So that replacing any one of them with a substituent results in the same product.
The second explanation is correct because benzene can be further substituted.
In 1874, Ladenburg proved experimentally that all the six hydrogen atoms in benzene are equivalent. Now structure III must be rejected, since it would form two isomeric monosubstituted derivatives as all the hydrogens in III are not equivalent. Moreover the structure contains four pi () bonds.
Similarly we can justify that structures II and IV are unsatisfactory. Thus, along with the Kekulé formula (I) , structure V is still a possibility.
5) Benzene yields only three isomeric disubstitution products, C6H4W2 or C6H4WZ.
When two hydrogen atoms in benzene are replaced by two univalent atoms or groups (which may be the same or different), three isomers are possible. Thus, three and only three isomeric dichlorobenzenes C6H4Cl2, and three bromonitrobenzenes, C6H4BrNO2, have every been found among the products.
On the basis of this fact, structure II, must be rejected.
Kelkule’s formula for benzene stimulated a large amount of research into the structure of aromatic compounds. Ladenburg (1869) attacked Kekulé’s formula on the grounds that it should give four disubstituted derivatives. At first glance, Kekulé’s formula (I) seems to be consistent with the fact that three and only three isomeric disubstituted benzenes are possible. For example, the three isomeric dichloro derivatives are the 1, 2-, the 1, 3-, and 1, 4- dichlorocompounds:
Closer examination of the Kekulés structure shows that, two 1, 2-dichloroisomers, differing in the positions of chlorine relative to the double bonds, should be possible.
In one of these hypothetical compounds (above), the carbon atoms that bear the chlorines are separated by a double bond, and in the other they are separated by a single bond. However, only one 1,2-dichlorobenzene has every been found.
Ladenburg therefore proposed his prism formula, (VII).
The six carbon atoms are at the corners of a regular prism, the edges of which denote linkages. This formula gives only one monosubstitution and only three disubstitution products. However, this formula does not contain any pi () bonds. It, therefore, fails to explain the addition reactions of benzene.
V. Meyer (1870) opposed Ladenburg by pointing out that the 1,2-and 1, 6-derivatives differed only in the position of double bonds and hence, he believed that, this difference would be too small to be noticeable practically. This way, V. Meyer supported Kekulés formula.
To accommodate the possible difference between the 1,2-and 1,6-positions, Kekulé proposed that the carbon atoms in benzene were continually in a state of vibration, and due to this vibration, each carbon-carbon pair had a double bond half of the time and the single bond the other half. This implied that there is an osciallation between the two forms (IX) and (X), each molecule spending half its time in (IX) and the other half in (X).
Thus, the benzene molecule is a dynamic thing. The two forms of benzene (and of benzene derivatives) are in a state of dynamic equilibrium and that this equilibrium is so rapidly established that it prevents isolation of the separate compounds. Thus, the two 1, 2-dichlorobenzenes would also be rapidly equilibrated and hence could not be separated.
Kekulés structure, thus, explains satisfactorily the facts (1), (2), (4), and (5). But the fact (3) is still not accounted for by this structure.
Kekulés structure of benzene appears to be “Cyclohexatriene”. One would expect this cyclohexatriene, like the cyclohexadiene and cyclohexene, to undergo readily the addition reactions characteristic of the alkene structure. As mentioned in the fact (3), in place of addition reactions, benzene readily undergoes a new set of reactions all involving substitution.
The tendency of benzene to react by substitution rather than addition leads to the concept of aromaticity. Thus, a compound should be called aromatic if it gives experimentally substitution reactions rather than addition reactions even though it is highly unsaturated.
Bayer proved that cyclohexane and hexahydrobenzene were identical, thus establishing the ring structure of benzene.
Bayer also observed that as soon as one double bond was removed from benzene, the “saturation’ properties were lost, and that dihydrobenzne (cyclohexadiene) behaved as would be expected of an alkadiene.
The outcome of Baeyer’s work was that aromatic properties’ depend on the peculiar symmetrical arrangement of the fourth valency of each carbon atom in the ring.
If aromatic character is due to the ring of alternating single and double bonds, a compound such as cyclooctatetraene should also exhibit ‘aromatic character’.
In 1911, Richard Willstatter synthesized cyclooctatetraene and performed chemical reactions on it. He found that it had typical unsaturated properties, that is, it is not at all like benzene. Cyclooctatetraene reacts with bromine by addition , it adds hydrogen readily, it is oxidized by solutions of potassium permanganate, and thus it is clearly not aromatic.
This led to a revival of Kekulé’s oscillation formulae. The difficulty with the Kekulé formula is that it represents benzene with three double bonds. We have seen that benzene shows unusual behavior by undergoing substitution reactions when, on the basis of its Kekulé structure, we expect it to undergo addition. This implies that benzene is unusual: It is more stable than the Kekulé structure suggests. Moreover, the oscillation does not account for the difference in behaviour between these and olefinic bonds.
6) Heats of hydrogenation and combustion of benzene are lower than expected Heat of hydrogenation (H0) is the amount of heat energy released when one mole of an unsaturated compound is hydrogenated. In almost all cases the value of H0 is about 28-30 kcal mol–1 for each double bond the compound contains.
Cyclohexene, a six-membered ring bearing one double bond, can be hydrogenated readily to cyclohexane.
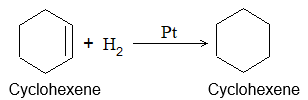
When the DH0 for this reaction is measured, it is not surprising that it is found to be -28.6 kcal mol–1 (-120 kJ mol–1), very much like that of any unsaturated compound containing one double bond.
Thus, one expects that hydrogenation of 1, 3-cyclohexadiene, a six-membered ring containing two double bonds, should liberated approximately twice as much heat and thus should have a H0 equal to about -57.2 kcal mol–1 (-240 kJ mol–1). The experimental value, H0 = – 55.4 kcal mol–1 (-232 kJ mol–1), is quite close to our calculation. The difference can be accounted by the fact that compounds containing conjugated double bonds are usually somewhat more stable than those that contain isolated double bonds:
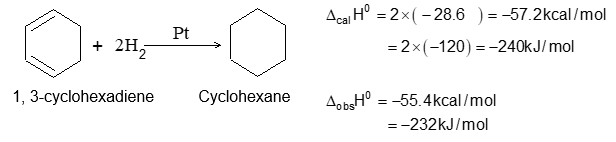
If benzene is simply 1, 3, 5-cyclohexatriene (hypothetical), we might reasonably expect benzene to liberate roughly three times as large as cyclohexene, that is, about 85.8 kcal mol–1 (or 360 kJ mol–1) when it is hydrogenated. The observed heat of hydrogenation of benzene is surprisingly different. It is -49.8 kcalmol–1 (or -208 kJ mol–1), that is 36 kcal (or 152 kJ) less than the expected amount.
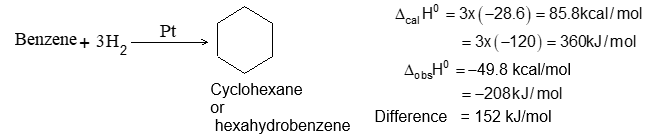
This entire quantitative approach can be more easily visualized with the help of a potential energy diagram, in which the height of a horizontal line corresponding to a particular molecule, represents the potential energy content of the molecule.
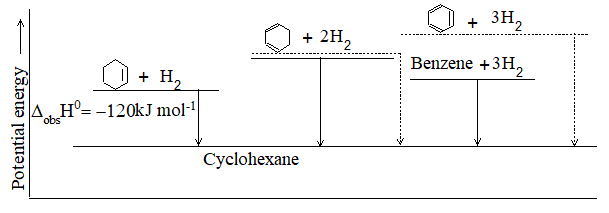
The final product cyclohexane in all the three cases is the same. The broken lines represent the expected results, based upon three equal steps of 28.6 kcal.
The fact that benzene releases 36 kcal (152 kJ) less energy than the hypothetical 1, 3, 5-cyclohexatriene can only imply that benzene contains 36 kcal (152 kJ) less energy than cyclohexatriene. In other words, with cyclohexatriene as the reference standard, benzene is more stable than this compound by 36 kcal (152 kJ). This value, difference between the amount of heat actually released and that calculated on the basis of the Kekulé structure hypothetical cyclohexatriene) is now called the resonance energy of the benzene.
Cyclohexene, and not ethene, is chosen as the reference compound to minimize any steric effects. The heat of combustion of benzene is also lower than predicted on the basis of the Kekulé structure, and by the same amount.
7) All carbon–carbon bonds in benzene are identical and have a length in between carbon–carbon single and carbon–carbon double bonds Spectroscopic measurements show that the molecule of benzene is a regular flat hexagon, (angle 1200) with all six hydrogen atoms lying in the plane of the ring and each C-C-H, C-C-C valency angle being 1200. They also show that all the six carbon – carbon bonds are of equal length viz., 1.39Å. This value lies in between that for a carbon-carbon single bond between sp2 – hybridized atoms (1.47 Å) and that for a carbon-carbon double bond (1.33 Å).
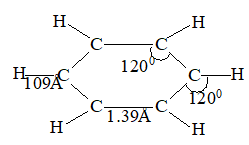
Thus, all the bonds in benzene have double-bond character.
Carbon-carbon double bonds in a wide variety of compounds are found to be about 1.33Å long. Carbon-carbon single bonds are considerably longer: 1.54Å in ethane, 1.50Å in propylene, and 1.47Å in 1, 3-butadiene.
If benzene actually possessed a Kekulé structure i.e., the hexagonal ring with three carbon-carbon single and three carbon-carbon double bonds, one would expect to find three short bonds (1.33Å) and three long bonds (1.48Å). This is completely against what we find experimentally.
The resonance view of the structure of benzene
One of the fundamental postulates of the theory of resonance (Quantum mechanical view) is that whenever more than one Lewis structure, differing only in the positions of their electrons, can be drawn for a molecule then none of the structures will represent the molecule completely and satisfactorily. With this concept of resonance, one can now understand the true character of the two Kekulé structures, I and II, for benzene.
The two Kekulé structures differ only in the arrangement of electrons. Therefore, they do not describe two separate molecules in equilibrium as Kekulé had proposed. Instead, they are the closest one can get to a structure for benzene within certain limitations.
The basic problem with the Kekulé structures is that they are typical Lewis structures which portray electrons in Localized distributions, but with benzene, the electrons are delocalized. Thus, the Kekulé structures I and II should be regarded as resonance contributors to the real picture of the benzene molecule. Thus, I and II should be connected with a double – headed arrow and not with two separate ones.
Resonance contributors are not in equilibrium.
They are not structures of real molecules. They are the closest we can get if we are bound by simple rules of valence, but they are very useful in helping us to visualize the actual molecule as a hybrid. Benzene is a hybrid of I and II. All of the C-C single bonds in structure I are C-C double bonds in structure II. If we blend I and II, that is, if we develop a hybrid of them, then all the carbon-carbon bonds in benzene will be identical and hence would have the some bond length. They are neither single bonds nor double bonds, rather, they have a bond order between that of a single bond and that of a double bond.
Actually there are five possible resonating structures. Of these two correspond to the Kekulé structures and the rest three to the Dewar structures. Calculations for weighing the contributions of the five hypothetical resonating structures show that the Kekulé structures contribute about 80 percent to the benzene resonance hybrid. For most purposes we use only the two Kekulé structures as the resonating forms of benzene.
‘Dewar benzene’ (bicyclo [2, 2, 0] hexadiene) has been prepared. It is not planar.
According to resonance theory, whenever equivalent resonance structures can be drawn for a molecule, the molecule (or hybrid) is much more stable than any of the resonance structures would be individually, if they could exist.
Since resonance contributors I and II are exactly identical, and hence of exactly the same stability they make equal contributions to the hybrid. And also, stabilization due to resonance should be large. In this way, resonance theory accounts for the much greater stability of benzene when compared to the hypothetical 1, 3, 5-cyclohexatriene, hence its aromatic character. For this reason, the extra stability associated with benzene is called its resonance energy.
The hybrid structure is represented by inscribing a circle in the regular hexagon, and it is this new formula (III) that is most often used for benzene today.
Formula III is a highly useful representation since it highlights the equivalence of the various carbons-carbon bonds.
It is understood that a hydrogen atom is attached to each angle of the hexagon unless another atom or group is indicated. The straight lines stand for the bonds joining carbon atoms. With benzene the circle represents the six electrons that are delocalized about the six carbon atoms of the benzene ring.
Another view point, the straight lines stand for single bonds, and the circle stands for the extra half-bonds.
With other aromatic systems, however, a circle in a ring may represent numbers of delocalized electrons other than six.
There are situations when an accounting of the electrons becomes essential. For such situations one may use one or the other of the Kekulé¢ structures because the electron count in a Kekulé¢ structure is obvious, whereas the number of electrons represented by a circle or portion of a circle is ambiguous. The presence of the circle distinguishes the benzene ring from the cyclohexane ring, which is generally represented today by a plain hexagon.
Any difficulty in accounting for the number of isomeric disubstituted products is sorted out by the fact that all carbon-carbon bonds in benzene are equivalent. It is now very clear that there can’t be more than three isomers, in agreement with experiment:
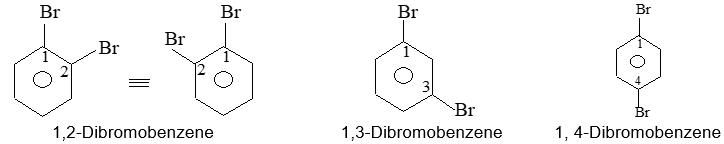
The “unusual” stability of benzene is not unusual at all: it is very much expected of a hybrid of equivalent structures. It is the 36 kcal of resonance energy that is responsible for its aromatic properties.
Alkenes readily undergo addition reactions because these reactions convert an alkene into a relatively more stable saturated compound. Thus hydrogenation of cyclohexene is accompanied by the release of heat, 28.6 kcal, as the product cyclohexane lies 28.6 kcal lower than the reactant on the energy scale.
But benzene resists addition, as addition would convert it into a less stable product by destroying the resonance-stabilized benzene ring system. Thus, benzene needs 5.6 kcal to form the less stable cyclohexadiene in the first stage of hydrogenation. Thus benzene prefers to undergo substitution in which the ring system is retained.
The molecular orbital view of the structure of benzene
Since the bond angles of the carbon atoms in the benzene ring (a regular flat hexagon) are all 1200, all the six carbon atoms in benzene are sp2 hybridized i.e., in a state of trigonal hybridization. These sp2 hybrid orbitals lie in the same plane, that of the carbon nucleus, and are directed, towards the corners of an equilateral triangle. To permit maximum overlap of the orbitals (sp2 H.O. of carbon and s orbitals of hydrogen), the six carbons and six hydrogens of benzene should be arranged to create the following structure.
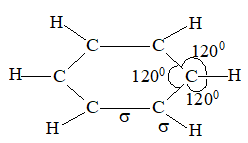
Two sp2 hybrid orbitals of each carbon atom overlap with sp2 hybrid orbitals of adjacent carbon atoms to form six C-C sigma bonds which are in the hexagonal plane. The remaining sp2 hybrid orbital of each carbon atom overlaps with s orbital of a hydrogen atom to form six C-H sigma bonds. Each bond orbital is cylindrically symmetrical about the line joining the atomic nuclei. Thus, these bonds are labeled as sigma () bonds.
The structure of the benzene molecule is still incomplete as there are six 2pz electrons to be considered. In addition to the three sp2 hybrid orbitals already used to form s bonds, each carbon atom has a fourth unhybridized 2pz orbital. This 2pz orbital consists of two equal lobes, one lying above and the other lying below the plane of the sp2 hybrid orbitals i.e. above and below the plane of the ring. It is occupied by a single electron.
Hence, in the incomplete structure of benzene there are six sigma C-H bonds, six sigma C-C bonds and six 2pz orbitals (one on each carbon atom) which are all parallel and perpendicular to the plane of the ring:
Like the case of an alkene, the p orbital of one carbon can overlap with the p orbital of its neighboring carbon, forming an additional pi () bond through the pairing of the electrons. But each carbon has a p orbital available for overlap with p orbitals of its adjacent carbons. Thus the overlap in benzene is not restricted to a pair of p—orbitals as it happens in an alkene. The p-orbital of any one carbon atom overlaps equally well with the p-orbitals of both carbons to which it is bonded. The favorable overlap of these p orbitals all around the ring results in a molecular orbital embracing all six carbon atoms, and encompassing the top and bottom faces of the ring. Therefore, all six 2pz electrons are completely delocalized resulting in two continuous doughnut-shaped electron clouds, one lying above and the other below the plane of the ring.

According to the theory of quantum mechanics, the number of molecular orbitals in a molecule is always equal to the number of atomic orbitals combined to form the molecule. Like an atomic orbital, each molecular orbital can accommodate a maximum of two electrons provided their spins are opposed. Right now, we consider only the p atomic orbitals contributed by the carbon atoms of benzene.
Since six 2pz atomic orbitals are involved, six pi () molecular orbitals are possible. Three of the molecular orbitals have energies lower than that of an isolated p orbital, these are the bonding molecular orbitals. The other three molecular orbitals have energies higher than that of an isolated p orbital, these are the antibonding molecular orbitals. These molecular orbitals are shown in the following figure:
(I), (II), (III) are bonding, and (IV), (V), (VI) are antibonding. MOs (II) and (III) have the same energy and are said to be degenerate; the same is true of MOs (IV) and (V). All MOs have a nodal plane in the plane of the ring. The lowest energy MO in benzene has overlap of p orbitals with the same mathematical phase sign all around the top and bottom faces of the ring. In this orbital there are no nodal planes (changes in orbital phase sign) perpendicular to the atoms of the ring. The MOs (II) and (III) of next higher energy level each have one nodal plane perpendicular to the plane of the ring. These orbitals are of equal energy (degenerate) because they both have one nodal plane. The next higher energy set of p MOs, (IV) and (V), each has two nodal planes perpendicular to the plane of the ring. These orbitals are also degenerate for the same reason. The highest energy MO of benzene, (VI), has three nodal planes perpendicular to the plane of the ring.
Since benzene is cyclic, instead of nodal points there are nodal planes (shown as lines) that are perpendicular to the plane of the ring.
In general, each set of higher energy MOs has an additional nodal plane.
The electronic configuration of the ground state of benzene is obtained by adding the six p electrons to the six p molecular orbitals, discussed above, starting with the orbitals of lowest energy.
Thus, in the ground state, the six 2pz electrons of benzene will occupy, in pairs, the MOs (I), (II) and (III). These are the MOs in which the number of nodal planes is fewer than in (IV), (V) and (VI). When benzene is in an excited state, one or more of the p-electrons will occupy the higher energy level MOs.
Since all of its bonding orbitals are filled with electrons having their spins paired, and no electrons are present in antibonding orbitals, benzene is said to possess a closed bonding shell of delocalized p electrons, accounting for the stability of benzene.
In the group state, the total energy of the three pairs of delocalized electrons is less than that benzene molecule is stabilized by delocalization i.e., resonance.
The electrons of benzene are not localized but are evenly distributed around the top face and bottom face of the carbon ring in benzene.
Crystalline benzene involves perpendicular interactions between benzene rings, so that the relatively positive periphery of one molecule associates with the relatively negative faces of the benzene molecules aligned above and below it.
The sum of three bonding MOs, holding the six electrons of benzene, is the symmetrical clouds above and below the plane of the ring.
It is overlap of the p orbitals in both directions and the resulting participation of each p electron in several bonds that corresponds to description of the benzene molecule as a resonance hybrid of two Kekulé structures.
Very much the same representation was advanced as long ago as 1899 by Johannes Thiele, who used a broken circle to stand for partial bonds i.e., partial valences.
Despite delocalization, the electrons of the benzene molecule are more loosely held than its electrons. Thus, these electrons become easily available to a reagent which seeks electrons. Hence, the typical reactions of the benzene ring are those in which it serves as a source of electrons for electrophilic or acidic reagents. Due to resonance stabilization, these reactions cause substitution, in which the aromatic character of the benzene ring is preserved.
The Hückel (4n + 2) rule for aromaticity
From the experimental standpoint, all aromatic compounds have the following properties in common:
1) Molecular formulae of aromatic compounds exhibit a high degree of unsaturation.
2) In spite of high degree of unsaturation, aromatic compounds are resistant to the addition reactions generally characteristic of unsaturated compounds.
3) Instead of addition reactions, aromatic compounds undergo electrophilic substitution reactions like those of benzene.
4) Aromatic compounds possess low heats of hydrogenation and low heats of combustion.
5) Aromatic compounds are cyclic – usually containing five -, six -, or seven – membered rings.
6) Aromatic compounds have flat (or nearly flat) molecules.
7) Protons of aromatic compounds show the same sort of chemical shift in NMR spectra as the protons of benzene and its derivatives.
Now, we discuss the necessary and essential requirement from a theroretical standpoint. In 1931, the German physicist Erich Hückel performed a series of mathematical calculations based on the theory of quantum mechanics. He carried out molecular orbital calculations on monocyclic systems CnHn containing n -electrons and each carbon atom providing one -electron His calculations show that planar monocyclic rings containing (4n + 2) electrons, where n = 0, 1, 2, 3, …and so on (i.e., rings containing 2, 6, 10, 14 ….., etc electrons), have closed shells of delocalized electrons like benzene and should have substantial delocalization energies or substantial resonance (stabilization) energies. In other words, planar monocyclic rings containing 2, 6, 10, 14, etc. delocalized electrons should be aromatic.
Thus Hückel’s rule applies to compounds containing one planar ring in which each atom is capable of being sp or sp2 hybridized, thus providing a p- orbital for extended p bonding in benzene.
Hückel’s rule states that to be aromatic a compound whether or not having benzene ring, must have a molecule that
i) is planar or reasonably planar,
ii) contains cyclic clouds of completely delocalized p electrons above and below the plane of the molecule, and
iii) contains a total of (4n + 2) – delocalized electrons in the clouds (n = 0,1,2,3, …. any integer)
In this description of aromaticity, no mention is made of the number of carbon atoms in the ring; the essential requirement is the presence of (4n + 2) -completely delocalized electrons, which is directly connected with the (reasonable) planarity of the ring. If the ring is not planar, favorable overlap of the p-orbitals is diminished or absent.
For the particular degree of stability that characterizes an aromatic compound, delocalization alone is not enough. There must be a specific number of electrons, called the Hückel number. Some Hückel numbers are 2, 6, 10, 14 and 18. This requirement has to do with the filling up of the various orbitals that make up the -cloud.
Polygon and circle rule developed by C.A. Coulson is a simple way to make a diagram of the relative energies of the molecular orbitals (obtained from Hückel’s calculations) of conjugated monocyclic systems. In this method, a regular polygon corresponding to the ring of the compound is inscribed in a circle with one vertex (corner) of the polygon placed at the bottom of the circle. For example, for benzene, the polygon is a regular hexagon. At the points where vertices of the polygon touch the circle, draw short horizontal lines outside the circle to indicate the relative energies of the molecular orbitals of the system. Now place a dashed horizontal line halfway of the circle to divide the bonding MOs from the antibonding MOs. The orbital falling on this line is a non bonding MO.
The vertices of the polygon touching the circle represent the relative energies of the MOs with energies increasing from the bottom to the top of the circle. The vertices below the horizontal diameter are bonding MOs, those above are antibonding * MOs, and these on the diameter are nonbonding n MOs.
Find the number of pi () electrons in the molecule. Place electron arrows on the lines representing the respective MOs, beginning at the lowest energy level and working upward. In doing so, Hunds’ rule must be followed to fill the degenerate orbitals i.e., fill each degenerate orbitals with one electron first and then add to each unpaired electron another with opposite spin if it is available.
Let’s apply the polygon rule to 3-, 4-, 5-, 7-, and 8- carbon systems: the following figure shows the energy levels and configurations of p electrons for monocyclic systems, CnHn, where n is 3-8, except 6. Note that the number of energy levels is equal to the number of carbon atoms and that MO energy levels which are the same for a given molecule are said to be degenerate. Inspection of figure, where all the molecules are in the ground state, shows that only benzene has (4n + 2) p-electron system (n = 1) and consequently is the only molecule which has a closed shell electron configuration. Thus, for the 3-8 rings, only benzene is aromatic.
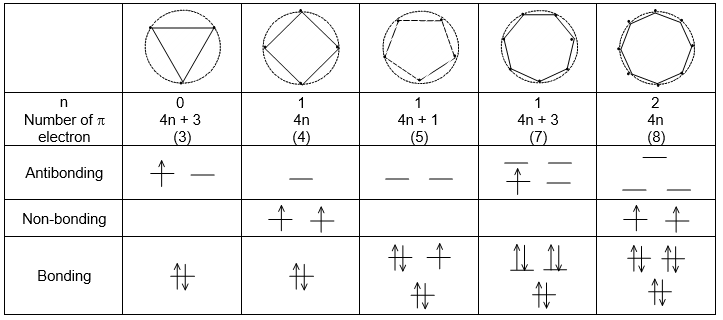
Illustration 23: Explain, why cyclooctatetracene is not aromatic?
Solution: Cyclooctatetreene has a total of eight electrons. Eight is not a Hückel number i.e. not a 4n + 2 number, it is a 4n number. Assuming cycloctatetraene to be planar, we can apply the polygen-and-circle method.
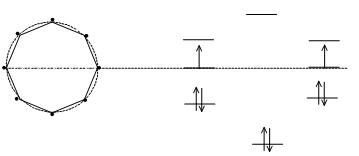
Like benzene, it does not have a closed shell of electrons. It has an unpaired electron in each of two nonbonding orbitals. Molecules with unpaired electrons (radicals) are not usually stable; they are typically highly reactive and unstable. A planar form of cyclooctatetraene is therefore, not at all like benzene and hence can’t be aromatic.
Because cyclooctatetraene does not gain stability by becoming planar, it assumes the tub shape:
There is relatively small strain in this conformation. If the double bonds were delocalized, the molecule would be flat and consequently would experience large strain, too large to be compensated by the gain in resonance energy.
The bonds of cyclooctatetraene are known to be alternately long and short, X-ray studies indicate that they are 1.48 and 1.34 Å, respectively.
Besides the neutral molecules discussed above, there are a number of monocyclic species that carry either a negative charge or a positive charge. Some of these ions showing unexpected stabilities seem to be aromatic. Hückel’s rule is quite helpful in accounting for their aromaticity.
Case I : Cyclopropenyl radical
having 3 pi () electrons is not aromatic
Aromatic as it has a doubly filled bonding and a singly occupied antibonding MO. If the odd electron is lost the resulting cyclopropenyl cation becomes a closed-shell (4n + 2) – electron species (n = 0), and hence an aromatic cation. It may be represented as a resonance hybrid (V.B. method):
Since there is an empty 2pz orbital, this can overlap with the two singly-occupied 2pz orbitals, to form a closed MO containing two p-delocalized electrons (M.O. method).
Many cyclopropenium salts have actually been prepared. These salts can be stored in a bottle.
Case II: Cyclobutadiene,

is a (4n) -electron molecule (n = 1). Its electronic
distribution in the ground state shows that it is in the triplet state because of the half-filled nonbonding MOs. This accounts for its high instability.
Cyclobutadiene has not yet been isolated, but the tetra-substituted derivative,
has been prepared.
All attempts to synthesize cyclobutadiene lead to the formation of
(i) a simple open chain molecule and products formed from it (Reverse cycloaddition).
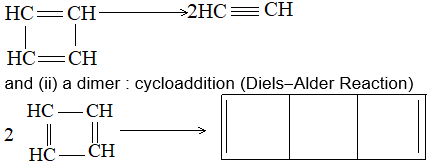
In solid argon, at a few degrees above absolute zero, cyclobutadiene has been generated and its structure determined. It has extra long C-C bonds to localize the C = C bonds and thereby minimize the extended – orbital overlap which induces the antiaromaticity (Molecules will always have structures with the lowest possible energies).
On the other hand, loss of two -electrons in the separate nonbonding MOs produces the closed-shell (4n + 2) -electron species (n = 0), the cyclobutenyl dication:
Tetraphenylcyclobutenyl fluoroborate has been prepared:
Gain of two -electrons in the separate nonbonding MOs also produces the closed shell (4n + 2) -electron species (n = 1), the cyclobutenyl dianion:
Thus, for four-membered rings to be aromatic, it is necessary to have a pAO on each C with a total of either two or six electrons for the overlapping system.
Case III Cyclopentadiene,
is not aromatic because it is a (4n) -electron molecule (n=1).
Also, cyclopentadienyl radical
is not aromatic because it is a (4n + 1)
-electron molecule (n = 1). However cyclopentadiene is unusually acidic though a typical hydrocarbon, indicating that loss of a hydrogen ion (H+) gives an unusually stable anion. [The pKa for cyclopentadiene is 15 to 16]. Because of its acidity, cyclopentadiene can be converted to its anion by treatment with moderately strong bases:

NMR spectroscopy shows that all five hydrogen atoms in the cyclopentadienyl anion are equivalent and absorb downfield.
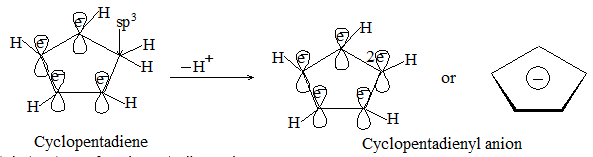
The orbital structure of cyclopentadiene shows
i) it does not have the proper number of p electrons,
ii) the electrons cannot be delocalized about the entire ring because of the intervening sp3-hybridized -CH2 – group with no available p-orbital.
This prevents cyclopentadiene from being aromatic.
After the lost of a proton, the -CH2– carbon atom can become sp2 hybridized and the two electrons left behind can occupy the new p orbital produced. Now, this new p orbital can overlap with the p orbitals on either side of it giving rise to a ring with six delocalized electrons. Since six is a Hückel number [4n + 2, where n = 1], the cyclopentadienyl anion is an aromatic anion. The unusual acidity of cyclopentadiene is a direct consequence of the unusual stability of its anion.
The gain of one electron by cyclopentadienyl free radical results in the formation of the cyclopentadienyl anion, which has three doubly filled bonding MOs.
Cyclopentadienyl anion is a closed-shell (4n + 2) – electron molecule (n = 1).
Pauson (1951) treated cyclopentadienyl magnesium bromide with ferric chloride and isolated dicyclopentadienyl iron:
This iron (Fe II) complex was named ferrocene by Woodward to indicate its aromatic character. It is a stable molecule that possesses a sandwich structure i.e., the two five-membered rings lie in parallel planes with the iron atom placed symmetrical between the two. It is also called the antiprism structure.
The molecule is symmetrical and therefore has a zero dipole moment. All of the C-H bonds are equivalent. All carbon-carbon bonds are 1.4Å. The entire ring is bonded uniformly to the metal atom. The bonding occurs by overlap of the sextet of -electrons of the ring with the d-orbitals of the metal, thereby giving a delocalized covalent bond between the metal atom and the cyclopentadienyl ring as a whole.
Ferrocene is an orange solid, m.p. 1730C, and its reactions are aromatic, e.g., it gives a mono-and diacetyl derivative by means of the Friedel-Crafts reaction under suitable conditions.
Illustration 24: Describe the electron distribution in the MOs of the cyclopentadienyl anion.
Solution: It has six (Hückel number) delocalized p electrons. Thus the electron distribution is:
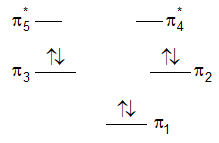
Illustration 25: Show that cyclopentadienyl cation is a diradical.
Solution: Cyclopentadienyl cation,
has four electrons. Its electronic distribution is :
The and bonding MOs are half-filled, thus, it is a diradical.
Case IV Cycloheptatriene,
commonly called tropylidene has six pi ()
electrons which cannot be fully delocalized because of the presence of the -CH2– group having no available p orbital. When cycloheptatriene is treated with a reagent capable of abstracting a hydride ion, it is converted to the cycloheptatrienyl (or tropylium) cation.
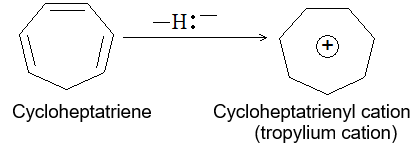
The loss of a hydride ion from cycloheptatriene occurs with unexpected ease, and the cycloheptatrienyl cation is found to be unusually stable. As a hydride ion is removed from the -CH2– group of cycloheptatriene, the carbon atom becomes sp2 hybridized and a vacant p orbital is created. Thus, the cycloheptatrienyl cation has seven overlapping p orbitals containing six delocalized electrons. This makes it an aromatic cation.
The NMR spectrum of the cycloheptatrienyl cation indicates that all seven hydrogen atoms are equivalent
Cycloheptatrienyl radical
has 7 electrons. By loss of the single p-electrons in the anti-boning MO gives the tropylium cation, a (4n+2) p-electron species (n=1).
Many tropylium (tropenium) salts have actually been prepared.
Illustration 26: Show the electron distribution in the molecular orbitals of the cycloheptatrienyl cation.
Solution: The polygon-and-circle method shows that the three lowest energy MOs are below the diameter and are bonding. The four highest energy ones are above the diameter and are antibonding MOs.
Tropylium bromide, C7H7Br, melts above 2000C, is soluble in water but insoluble in non-polar solvents and gives an immediately precipitate of AgBr when treated with silver nitrate. This, strange behavior for an organic bromide, strongly suggests that, even in the solid, we are dealing with an ionic compound, R+Br–, whose cation is a stable carbocation.
Aromatic, nonaromatic and antiaromatic compounds
When we say that a given compound is aromatic we mean that its electrons are delocalized over the entire cyclic structure (ring) and that it is stabilized by the -electron delocalization.
NMR spectroscopy provides the best way and direct physical evidence of whether or not the electrons are delocalized.
When we say that a compound such as benzene is stabilized by -electron delocalization we mean that its heat of hydrogenation is much less than the heat of hydrogenation of the hypothetical 1,3,5-cyclohexatriene – a model in which the electrons are not delocalized. The energy difference between them is called the resonance energy or delocalization energy or stabilization energy. Thus, benzene in which electrons are delocalized is much more stable than the hypothetical 1, 3, 5-cyclohexatriene in which the electrons are not delocalized.
In order to make similar comparisons for other aromatic compounds we compare the -electron energy of the cyclic system with that of the corresponding open-chain compound. This approach is particularly useful because it furnishes us with models not only for annulenes but for aromatic cations and anions as well. However, when the cyclic system is strained, corrections are made.
In this approach, we take a linear chain of sp2 hybridized atoms that carries the same number of -electrons as our cyclic compound, the model for comparison. Next we imagine removal of two hydrogen atoms from the ends of this chain and joining the ends to form a ring. If the resulting ring has lower -electron energy than the open chain, then the ring is aromatic. If the ring and chain have the same -electron energy, then the ring is nonaromatic. If the ring has higher (greater) energy than the open chain, then the ring is antiaromatic.
Planar conjugated carbocyclic polyenes that are especially more stable than their open chain analogs are called aromatic. They possess (4n + 2) electrons; e.g., 2, 6, 10 …
Planar conjugated carbocyclic polyenes that are especially less stable than their open chain analogs are called antiaromatic. They posses (4n) electrons; e.g., 4, 8, …
Planar conjugated carbocyclic polyenes whose stability is comparable to their open chain analogs are called nonaromatic. They possess (4n + 2) electrons but a completely cyclic overlapping system is missing.
The following examples illustrate how this approach is used.
Cyclobutadiene: Consider the change in -electron energy for the following hypothetical transformation:
Both experiments and calculations confirm that the -electron energy of cyclobutadiene is higher than that of its open-chain counterpart. Thus cyclobutadiene is labeled as antiaromatic.
Cyclopentadienyl anion Using a linear anion for the following hypothetical transformation:
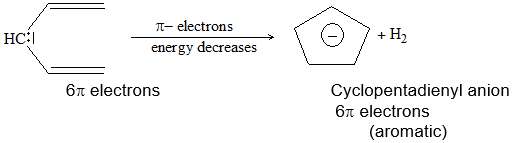
Calculations indicate and experiments confirm that the cyclic anion has a lower p-electron energy than its open-chain counterpart. Therefore, the cyclopentadienyl anion is classified as aromatic.
Benzene Considering the following hypothetical transformation for comparison:
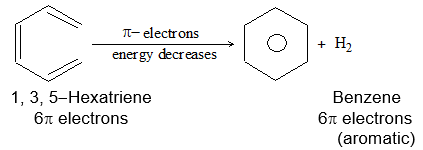
Both calculations and experiments confirm that benzene has a much lower -electron energy than 1, 3,4-hexatriene. Thus, benzene is designated as aromatic.
Cycloheptatriene
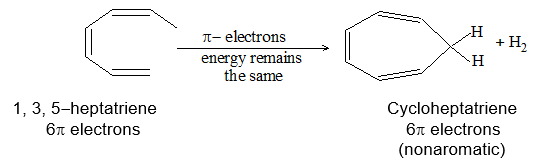
Here calculations and experiments indicate that the p-electron energy of cycloheptatriene is as same as that of its open-chain counterpart. Thus, cycloheptatriene is classified as nonaromatic.
One of the C’s of cycloheptatriene is sp3 hybridized, thereby preventing a completely cyclic overlapping system.
The same approach shows that 3-Methylene-1, 4cyclohexadiene,
is nonaromatic.
Cyclooctatetraene Consider the following hypothetical transformation:
Both calculations and experiments indicate that a planar cyclooctatetraene would have higher p-electron energy than the open-chain octatetraene. Therefore, a planar form of cyclooctatetraene would (if it existed) be antiaromatic. However, cyclooctatetraene is not planar and behaves like a simple cyclic polyene.
One can write Kekulé-type resonance structures for cyclooctatetraene:

Since these are equivalent energy resonance forms like those for benzene, one may predict cyclooctatetraene to be aromatic. However, the DH for hydrogenation of cyclooctatetraene in to cyclooctane is about four times the value than that of the addition of H2 to cyclooctene incidating that there is little or no electron delocalization (resonance). This implies that it behaves like a simple cyclic polyene.
Thus, the ability to write equivalent resonance contributors is not always reliable for predicting aromaticity of conjugated carbocyclic polyenes. MO theory, including Hückel’s rule, is the best method for predicting aromaticity.
Aromaticity is observed when all bonding MOs are completely filled (e.g. benzene) and nonbonding MOs if present, are either empty or completely filled. Hückel proposed his rule because of this requirement.
A species is antiaromatic if it has e–’s in antibonding MOs or if it has half-filled bonding MOs or nonbonding MOs, provided it is planar.
Illustration 27: Using MO theory, show that cyclooctatetraene is antiaromatic.
Solution: It is a (4n) -electron molecule. In the ground state the eight p electrons of cycloorctatetraene can be arranged in the MOs as shown:
Since the nonbonding MOs are half filled, a planar form cyclooctatetraene would, if it existed, be antiaromatic. Note that, it is in the triplet state, this accounts for its high (olefinic) reactivity.
Cyclooctatetraene undergoes reactions typical of alkenes-addition of electrophilic reagents like Br2 and HCl, oxidation by KMnO4 etc.
Cyclooctatetraene is not planar, it is actually tub-shaped:
The p-orbitals of one C = C are not coplanar with those of a neighboring C = C. Thus no effective overlap for delocalization can be possible. This non-coplanarity avoids the antiaromaticity which requires co-planarity. Thus, non-planar cyclooctatetraene is non aromatic.
(4n) -electron species do not possess a closed-shell planar configuration in the ground state. This is the reason for the instability or reactivity of these species. In fact, monocyclic (4n) -electron species are less stable than their acyclic analogues. This decreased stability in (4n) molecules has been called anti-aromaticity.
Gain of two pi () electrons (one to each nonbonding MO) by cyclooctatetraene gives the cyclooctatetraenyl dianion. It is a closed-shell (4n + 2) -electron molecule (n = 2), and hence aromatic:
Illustration 28: Using M.O. theory explain why cyclobutadiene is antiaromatic. Solution: The M.O. electron distribution is
Since the non-bonding MOs are half-filled, cyclobutadiene is antiaromatic.
The Annulenes
Annulene is a general term for conjugated monocyclic polyenes i.e., monocyclic compounds having alternating single and double bonds. The ring size of an annulene is indicated by a number in brackets, Hückel’s rule predicts that annulenes will be classified as aromatic, provided their molecules have a planar carbon skeleton and have (4n + 2) -delocalized electrons.
Before 1962, the only conjugated monocyclic polyenes that were available to test Hückel’s predictions were benzene ([6] annulene) and cyclooctatetraene ([8] annulene). During 1962, F. Sondheimer synthesized a number of large-ring annulenes to test the Hückel’s (4n + 2) rule. Their aromatic character was verified by NMR spectroscopy. Thus, annulene names are often used for conjugated rings of 10 or more carbon atoms, but they are seldom used for benzene and cyclooctatetraene.
Conjugated monocyclic polyenes, CnHn, in which , are usually called annulenes. The annulenes prepared by F. Sondheimer have n = 12, 14, 16, 18, 20, 22, 24 and 30. Of these, only the [14], – [18]-, [22]- and [30] annulenes are (4n + 2) p electron molecules (n = 3, 4, 5,7 respectively). As Hückel’s rule predicts, they have been found to be aromatic. The rest are not aromatic. They are (4n) p molecules, not (4n + 2) molecules. They have olefinic properties as found in the case of cyclooctatetraene.
Polycyclic Benzenoid aromatic hydrocarbons
All of these aromatic compounds consist of molecules containing two or more benzene rings fused together. For example, naphthalene:
So far we have applied Hückel’s (4n + 2) rule to monocyclic systems. However, the rule is not highly successful for polycyclic conjugated systems as it applies in some cases, e.g., naphthalene (10 p – electrons), but not in others, e.g., pyrene (16 p -electrons). Both of these are aromatic chemically i.e., both are benzenoid compounds.
There is another set of polycyclic benzenoid aromatic hydrocarbons in which the benzene rings are isolated e.g., biphenyl
Physico-chemical evidence such as heat of combustion indicates that a molecule of naphthalene can be considered to be a resonance hybrid of mainly three Kekulé’s structures (resonance theory):
There are (n + 1) principal resonating structures for a polynuclear hydrocarbon containing n benzene rings fused together in a linear manner.
The most stable resonance contributor of a polynuclear aromatic compound is that which has the maximum number of rings in the benzenoid condition, i.e., three double bonds in each individual ring’. Thus, according to the Fries rule, naphthalene tends to behave as structure I as it has two benzenoid rings.
Let’s consider the most important Kekulé’ structure of naphthalene:
C4a and C8a, are two carbon atoms in naphthalene, that are common to both rings. These two atoms, at the points of ring fusion, direct all of their bonds toward other carbon atoms and do not carry hydrogen atoms.
Every carbon of naphthalene is sp2 hybridized and carries a p-orbital perpendicular to the plane of the ring. Each of these p-orbitals carries one e–.
The p orbitals overlap around the periphery of both rings and across the points of ring fusion.
Molecular orbital calculations indicate that delocalization of the 10 electrons over the two rings yields a structure (resonance hybrid) with considerably lower energy than that calculated for any individual Kekulé’ structure. Consequently, naphthalene has a substantial resonance energy.
Since two benzene rings are present, one expects that the resonance energy (RE) of naphthalene should be close to 2 150.6 = 301.2 kJ/mol. But RE of naphthalene is actually less, it is 255.2kJ/mol. Hence naphthalene is less aromatic than benzene as each ring in naphthalene has RE of 127.6 kJ.
Consequently, naphthalene will be more reactive than benzene, as it has less aromatic character than benzene.
Anthracene (C14H10) and phenanthrene (C14H10) are isomers. In anthracene the three rings are fused in a linear way while in phenanthrene they are fused in an angular way.
Both of these molecules possess large resonance energies and exhibit chemical properties typical of aromatic compounds.
Anthracene is best regarded as a resonance hybrid of four resonating structures of which the most important one (Fries rule) is:

Like naphthalene, anthracene is also less aromatic than benzene as it has a resonance energy of 351.5 kJ/mol.
The most important resonating structure for phenanthrene is (Fries rule) is :
Since its resonance energy is 387.0 kJ/mol, it is less aromatic than benzene but more aromatic than anthracene.
The formula of phenanthrene may be written in the three ways:
Let us consider the Kekulé structure for pyrene, an aromatic compound:
The total number of electrons in pyrene is 16:8 double bonds = 2 8 (16) electrons. Sixteen is not a Hückel number.
Hückel’s rule is basically concerned with compound containing one planar ring in which each atom provides a overlapping p orbital as in benzene, but pyrene is not monocyclic, rather clearly tetracyclic. If we ignore the internal double bond of pyrene and focus only at the periphery, we notice that the periphery is a planar monocyclic ring with 14 – electrons. Fourteen is a Hückel number (4n + 2, where n = 3), thus, in the absence of the internal double bond, the periphery of pyrene is predicted to be aromatic by itself.
This prediction was confirmed when V. Boekelheide synthesized trans-15, 16- dimethyl dihyropyrene and showed that it is aromatic.
Thus, it is suggested that the Hückel’s (4n + 2) rule should be applied to the peripheral (conjugated) p -electrons of polycyclic systems.
Non-benzenoid aromatic compounds
Conjugated cyclic systems (monocyclic as well as polycyclic) which are (4n + 2) p-electron molecules but do not carry fused or isolated benzene rings are referred to as non-benzenoid aromatic compounds.
Naphthalene, phenanthrene, and anthracene are examples of benzenoid aromatic compounds while the cyclopropenyl cation, the cyclobutenyl dication, the cyclopentadienyl anion, the cycloheptatrienyl cation, the aromatic annulenes and trans-15, 16 – dimethyldihydropyrene are classified as non-benzenoid aromatic compounds.
Another interesting example of a non-benzenoid aromatic hydrocarbon is the compound azulene, C10H8. At room temperature, it is an intensely blue solid, with a melting point of 990C. Azulene has a resonance energy of 205 kJ mol–1‑. It is a resonance hybrid of the following contributors:
There are two Kekulé resonating structures containing 10 -electrons [(4n + 2), n = 2, 10 peripheral electrons]. The five membered ring has five and the seven – membered ring has seven -electrons, the two -electrons are common to both rings.
Although neither the cyclopentadiene nor the cycloheptatriene ring alone is aromatic, if one -electron is transferred from the seven- to the five-ring, each ring will have a closed shell of six -electrons as in naphthalene.
Charged resonance structures show a predominance of negative charge in the five-membered ring, making it similar to the aromatic cyclopentadienyl anion. In these structures, the seven-membered ring bears a positive charge, imparting it the aromatic character of the cycloheptatrienyl cation.
The main contributing structures to the hybrid carry charge separation accounting for the appreciable dipole moment, 1.0 D. The molecule of azulene, therefore, will have a dipolar structure:
Chemically, azulene behaves as an aromatic compound, e.g. it undergoes bromination, nitration, and the Friedel-crafts reaction. Electrophilic substitution occurring preferentially in the five-membered ring as it is more electron rich than the seven-membered ring.
Fullerenes
One may regard diamond as the polycyclic aliphatic system and graphite as the fused-ring aromatic system. X-ray analysis shows that, in graphite, the carbon atoms are arranged in layers. Each layer is a continuous network of planar, hexagonal rings. The carbon atoms within every layer are held together by strong, covalent bonds 1.42Å long. They are only slightly longer than those in benzene, 1.39Å.
A third, relatively newly discovered (1985) allotrope of carbon is Buckminsterfullerene, named after the architect Buckminster Fuller, renowned for his development of structures with geodesic domes.
Unlike diamond and graphite, whose molecules go on and on, the fullerene has a definite formula: C60. The structure of C60 and its existence was established in 1985 by H.W. Kroto, R.E. Smalley, and R.F. Curl, and their co-workers. They had found both C60 and C70 as highly stable components of a mixture of carbon clusters formed by laser-vaporizing graphite. In 1990 W. Krafschmer, D. Huffman and their co-workers, described the first practical synthesis of C60 by the resistive heating of graphite in an inert atmosphere. C60, a molecule shaped like a soccer ball, is a member of an exciting new group of aromatic compounds called fullerenes.
Fullerenes are cage like molecules with the geometry of a truncated icosahedron or geodesic dome. Like a geodesic dome, a fullerene is composed of a network of pentagons and hexagons. To close into a spheroid, a fullerene must have exactly 12 five-membered, but the number of six-membered faces can vary widely. In C60 molecule, the fused-ring aromatic system bends around and closes to form a soccer ball-shaped molecule (a ‘bucky-ball’) with 20 six-membered hexagonal rings and 12 five-membered rings. It has been called the “most symmetrical possible molecule”.
Since 1990 chemists have synthesized many higher and lower fullerenes. C70 having 25 hexagonal rings is elongated like a rugby ball. R.E. Smalley predicts that a “Russian doll” molecule can be made –it is a C60 molecule trapped inside a very large (C240) fullerene.
Each carbon of a fullerene is sp2 hybridized and forms bonds to three other carbon atoms. The remaining p electron at every carbon is delocalized into a system of molecular orbitals that gives the whole molecule aromatic character. Chemistry of fullerenes is quite fascinating: there is room inside the hollow balls for metal ions; the outer surface can be modified by chemical reactions.
Fullerenes have a high electron affinity. Thus, they readily accept electrons from alkali metals to produce a new metallic phase – a “buckide” salt. One such salt K3C60, is a stable metallic crystal consisting of a face-centered cubic structure of ‘buckyballs” with a potassium atom in between. It becomes a superconductor (i.e., offering resistance free conductivity of electricity) when cooled below 18 K. Therefore, compounds of fullerenes offer the prospect of superconductivity not at the very low temperatures so far required but at room temperature. Fullerenes have even been synthesized that have metal atoms in the interior of the carbon atom cage.
Heterocyclic Aromatic compounds
In almost all of the cyclic compounds that we have discussed so far – benzene, naphthalene, cyclopentadiene – the rings are composed entirely of carbon atoms. Such compounds are called Homocyclic or carbocyclic compounds. But in molecules of many cyclic compounds an element other than carbon is present in the ring. These compounds are called heterocyclic compounds. Thus, heterocyclic compounds are cyclic compounds that contain a ring made up of more than one kind of atom i.e., carbon and other elements, the commonest being nitrogen, oxygen or sulphur.
There are a number of heterocyclic rings which are easily opened and do not possess any aromatic properties, e.g., ethylene oxide, g- and d-lactones, etc. These are, usually, not considered to be heterocyclic compounds. Truly speaking, heterocycles are those compounds with five- or six- membered heterocyclic rings which are stable, contain conjugated double bonds, and exhibit aromatic character. They are quite commonly encountered in nature.
Four most common examples of heterocyclic compounds are shown here in their Kekulé’ forms:
All these four compounds are aromatic. Examining these structures, one notices that pyridine is electronically related to benzene, and that pyrrole, furan and thiophene are related to the cyclopentadienyl anion.
Pyridine is flat, with bond angles of 1200. The four carbon-carbon bonds have the same length. Similarly, the two carbon-nitrogen bonds are of the same length. It resists addition and undergoes electrophilic substitution. It has a resonance energy of about 23 k cal mol–1 (or 125.5 kJ/mol), as indicated by its heat of combustion. Pyridine can be regarded a resonance hybrid of the two Kekulé’ structures I and II. The hybrid can be represented as III, in which the circle stands for the aromatic sextet:
The nitrogen atom in pyridine is sp2 hybridized. Thus the nitrogen atom, like each of the carbon atoms, is bonded to other members of the ring by the use of sp2 orbitals, and donates one bonding electron to the p electron cloud. This electron, together with one from each of the five carbon atoms, gives pyridine a sextet of electrons like benzene. The third sp2 orbital of each carbon atom is used to form a bond to hydrogen. The two unshared electrons of the nitrogen atom of pyridine are in an sp2 hybrid orbital. It lies in the same plane as the atoms of the ring. This sp2 orbital does not overlap with the orbitals of the ring. Thus, it is said to be orthogonal to the orbitals. The unshared electron pair on nitrogen atom is not a part of the system. Thus, these electrons are available for sharing with acids, conferring on pyridine the properties of a (weak) base.
Pyrrole is a very important five-membered heterocyclic ring because its nucleus occurs in many natural compounds e.g., alkaloids, chlorophyll, haematin, etc.
Each atom whether carbon or nitrogen, of the pyrrole ring is attached to three other atoms by a bond. To form these bonds, the atom makes use of three sp2 hybrid orbitals. These orbitals lie in a plane and are 1200 apart. After contributing one electron to each of the three sigma () bonds, each carbon atom of the ring is left with one electron and the nitrogen atom is left with two electrons.
All these electrons occupy p-orbitals. Overlap of these p orbitals gives rise to pi () clouds, one above and one below the plane of the ring. These clouds contain a total of six delocalized electrons, the aromatic sextet. Delocalization of the electrons stabilizes the ring of pyrrole. Consequently, pyrrole has an abnormally low heat of combustion.
Instead of addition, it tends to undergo substitution reactions in which the stabilized ring structure is retained. Nitrogen’s pair of electrons, which is responsible for the usual basicity of nitrogen compounds, is involved in the cloud and is part of the aromatic sextet. Thus, these electrons are not available for donation to a proton from an acid. Consequently, in contrast to most amines, pyrrole is an extremely weak base (Kb ~ 2.5 10-14) in aqueous solution. Since the sp2 –hybridized nitrogen contributes two electrons to give an aromatic sextet; there is a high electron density in the pyrrole’s ring. This makes pyrrole to be extremely reactive towards electrophilic substitution. For example, it undergoes reactions like nitrosation and coupling with the diazonium salts which are characteristic of only the most reactive benzene derivatives, phenols and amines.
Pyrrole can be considered as a resonance hybrid of the following contributors.
Donation of electrons to the ring by nitrogen is indicated by the ionic structures in which nitrogen bears a positive charge and the carbon atoms of the ring bear a negative charge. The resonance hybrid is represented as follows wherein the circle represents the aromatic sextet.
Furan and thiophene are structurally quite similar to pyrrole. The oxygen atom in furan and the sulphur atom in thiophene are sp2 hybridized.
In both compounds the p orbital of the heteroatom donates two electrons to the cloud, to give an aromatic sextet. As a result, these compounds behave like extremely reactive benzene derivatives. The oxygen and sulphur atoms of furan and thiophene carry an unshared pair of electrons in an sp2 orbital that is orthogonal to the system.
The estimation of the ring current in pyrrole shows that it is less aromatic than thiophene but more aromatic than furan.
Concept of aromaticity
Aromatic character (aromaticity) can be defined in different ways:
a) High degree of unsaturation
b) ‘Unusual’ chemical properties such as substitution rather than addition
c) Usual stability reflected by large resonance or delocalization energy
d) A (reasonably) planar cyclic structure with (4n + 2) p-delocalized electrons
e) If polycyclic, then it should have (4n + 2) p-delocalized peripheral electrons
Thus, a monocyclic compound is classified as aromatic if it contains a (reasonably) planar cyclic structure, carries (4n + 2) -delocalized electrons and has unusual stability.
This definition of aromaticity includes many compounds which have very little resemblance chemically to benzene.
Arenes
Benzene derivatives in which the substituents are alkyl, alkenyl or alkynyl groups are known collectively and generally as arenes. Thus, arenes are benzene homologues or aromatic hydrocarbons.
Arenes also include the alkyl etc., derivatives of polynuclear aromatics such as naphthalene, anthracene, etc.
An aryl group is derived from an arene by removal of a hydrogen atom. Its symbol is Ar-. Thus, arenes are designated ArH just as alkanes are designated RH.
Physical properties In general, arenes resemble other hydrocarbons in their physical properties.
1) In the absence of polar substituents, intermolecular forces are weak and limited to van der Waals attractions of the induced-dipole/induced-dipole type.
2) They are usually colourless liquids or solids with a characteristic aroma.
Naphthalene balls are used in toilets and for preservation of clothes because of unique smell of the compound and the moth repellent property.
3) Their boiling points rise fairly regularly with increasing molecular weight. The increment is usually 20-30 degrees for each CH2 In general, the boiling points of a group of isomers are fairly close.
4) They are non-polar or weakly polar.
5) They are less dense than water.
6) They are immiscible with water but are readily miscible with organic solvents like ether, carbon tetrachloride or ligroin.
7) They are inflammable, burning with a sooty (smoky) flame.
Chemical properties In spite of high degree of unsaturation, the characteristic reactions of benzene are substitution reaction in which the resonance – stabilized ring system is preserved.
There is a cloud of -electrons above and below the plane of the benzene ring. On account of resonance (delocalization), pi () electrons of benzene are more involved in keeping together carbon nuclei relative to the pi () electrons of the carbon-carbon double bond.
However, with respect to the sigma () electrons these pi () electrons are loosely held and are easily available to a reagent that is in the need of electrons. Thus, in its typical reactions, the benzene ring acts as a source of electrons, that is, as a (Lewis) base.
The typical reactions of the alkenes are electrophilic addition reactions while the typical reactions of the benzene ring are electrophilic substitution reactions.
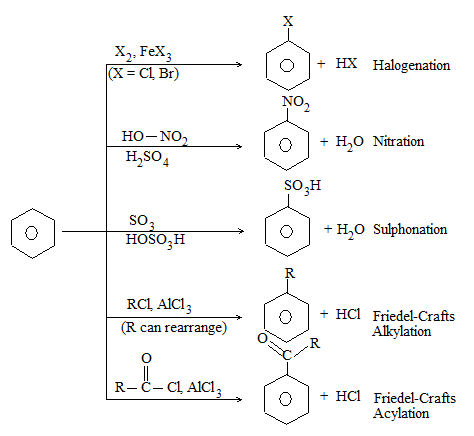
The important substitution reactions of benzene are summarized in the flow chart as follows:
These reactions are also the characteristic reactions of the benzene derivatives and of many aromatic rings, benzenoid as well as non-benzenoid.
Thus, the most characteristic reactions of benzenoid arenes are the substitution reactions that they undergo when they react with electrophilic reagents. These reactions are generally described as:
The electrophiles are either positive ions or neutral but electron deficient species with a large partial positive charge on a particular atom. For example, benzene can be nitrated when it reacts with nitric acid in the presence of sulphuric acid. The two acids react to produce positive nitronium ions, NO2+.
These positive nitronium ions act as electrophiles and attack the benzene ring, replacing one of the hydrogen atoms resulting in a reaction called an electrophilic aromatic substitution (EAS).
Electrophilic aromatic substitutions allow the direct introduction of a wide variety of atoms (or groups) onto an aromatic ring, thereby, providing synthetic routes to many important compounds.
EAS includes a wide variety of reactions:
a) halogenation, nitration, sulphonation, and Friedel-Crafts reactions, undergone by nearly all aromatic rings
b) reactions like diazo coupling and nitrosation, undergone only by rings of high reactivity
c) reactions like desulphonation, isotopic exchange, and many ring closures
Electrophilic Aromatic Substitution
1) Halogenation: Substitution of one of the ring hydrogens by a halogen atom:
2) Nitration: Substitution of one of the ring hydrogens by a nitro group:
3) Sulphonation: Substitution of one of the ring hydrogens by a sulphonic acid group:
4) Friedel–Crafts alkylation: Substitution of one of the ring hydrogens by an alkyl group:
5) Friedel–Crafts acylation: Substitution of one of the ring hydrogens by an acyl group :
6) Nitrosation: Substitution of one of the ring hydrogens by a nitroso (-NO) group. It is only for highly reactive ArH:
7) Diazo coupling: It is only for highly reactive ArH:
8) Kolbe reaction; It is only for phenols.
9) Reimer–Tiemann reaction: It is only for phenols.
10) Protonation : It involves desulphonation and hydrogen exchange:
.
Mechanism of Aromatic Substitution
There are three possible mechanisms for aromatic substitution: electrophilic, nucleophilic and free-radical. Of these, the most common substitution reactions involve electrophilic reagents.
Benzene is susceptible to electrophilic attack mainly because of its exposed pi (p) electrons. This way benzene resembles an alkene, because when an alkene reacts with an electrophile, the site of attack is also the exposed bond.
However, benzene differs from an alkene significantly. Benzene’s closed shell of six pi () electrons imparts it a special stability. Thus, although benzene is susceptible to electrophilic attack it undergoes substitution reactions rather than addition reactions, because, in substitution reactions, the aromatic sextet of pi () electrons is regenerated after attack by the electrophile.
Electrophilic aromatic substitution (EAS) reactions proceed by a single mechanism, whatever the particular reagent involved. The widely held theory of electrophilic aromatic substitution is that it proceeds by a bimolecular mechanism via the formation of an intermediate, and that the formation of this intermediate is the rate-determining step.
In each case there is a preliminary acid-base reaction which generates the attacking particle. However, the actual substitution is contained in two steps only.
In step 1 the electrophilic attacks the p cloud of benzene to form a non-aromatic cyclohexadienyl carbocation (benzenonium cation) known generally as an arenium ion. The entering group (electrophile) approaches the ring in a lateral direction with respect to the plane of the ring:
Step 1
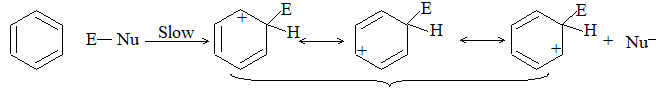
In illustrating mechanism, it is convenient to use Kekulé’ structures, because these make it much easier to keep track of the electrons.
Resonance structures such as those used for the arenium ion are important for study of electrophilic aromatic substitution.
In first step the attacking electrophile forms a sigma () bond to one of the carbon atoms of the ring by taking two electrons of the aromatic sextet (six-electron p system). In the formation of the arenium ion the carbon atom that forms a bond to the electrophile changes its state of hybridization from trigonal (sp2 to tetrahedral (sp3) and, therefore, no longer carries an available p orbital. Thus, formation of this bond destroys the cyclic system of electrons as the attacked carbon is removed from conjugation with the rest of the system. The arenium ion although no longer benzenoid, still has a conjugated system which exhibits resonance. Five carbon atoms of the ring are still sp2 hybridized and still have p orbitals available. The four p electrons of the arenium ion are delocalized through these five p orbitals.
In step 2 a hydrogen atom from the carbon (of the arenium ion) that bears the electrophile is removed as proton. The two electrons that bond this proton to carbon now become a part of the cloud (system). The carbon atom that carries the electrophile again becomes sp2 hybridized. This way, a benzene derivative with six fully delocalized electrons, is formed. Describing step 2 with any one of the resonance structures for the arenium ion gives:
Step 2
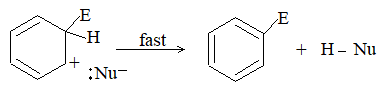
To show the mechanism using modern formula for benzene we can do it the following way:
Step 1 :
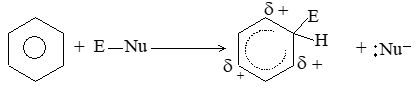
Step 2:
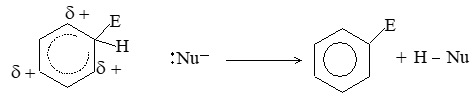
Here we have drawn the arenium ion as a delocalized cyclohexadienyl cation. For this purpose, the three resonating structures contributing to the intermediate are combined.
Notice that the o- and p- positions carry small positive charges.
There is firm experimental evidence that the arenium ion is a true intermediate in electrophilic aromatic substitution reactions. It is not a transition state.
A more detailed picture of this mechanism is shown in a free-energy diagram:
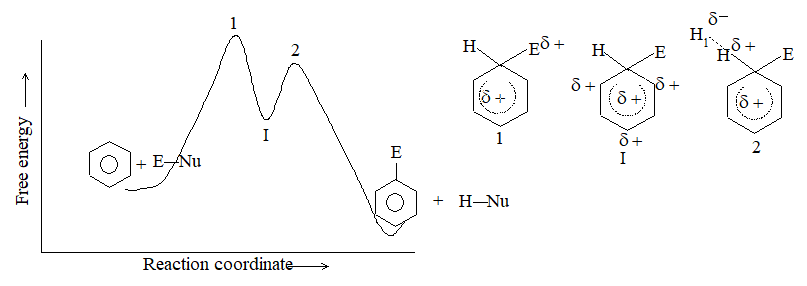
Notice that in a free-energy diagram the arenium ion lies in an energy valley between two transition states 1 and 2. The arenium ion is preceded by the formation of its transition state from the reactants. In this transition state 1, the bond between the electrophile and one carbon atom of the benzene ring is only partially formed. The arenium ion now produces the products via the transition state (T.S.)-2. In T.S.-2 the bond between the same benzene carbon atom and its hydrogen is partially broken. The bond between the hydrogen atom and the conjugate base (:Nu–) is partially formed.
Notice that the free energy of activation, , for the reaction leading from benzene and the electrophile, E+, to the arenium ion is much higher (greater) than the free energy of activation, , for the reaction leading from the arenium ion to the final product.
Also note that, the reaction leading from benzene and an electrophile to the arenium ion is highly endothermic, because the benzene ring loses its resonance energy. By contrast, the reaction leading from the arenium ion to the substituted benzene is highly exothermic because in it the benzene ring regains its resonance energy.
Of the following two steps, step 1 (the formation of the arenium ion) is usually the slow and hence the rate-determining step in electrophilic aromatic substitution. Step 2, the removal of a proton takes place rapidly relative to step 1. It has no effect on the overall rate of reaction. Accepting that substitution is electrophilic, how do we ensure that electrophilic aromatic substitution involves two steps instead of just one involving synchronous formation of the C-E bond and breaking of the C-H bond.
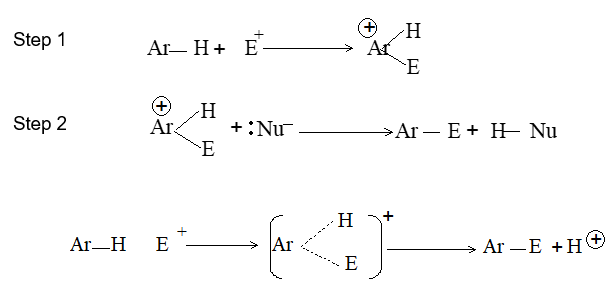
Moreover, how do we know that in the two-step mechanism, first one is much slower than the second, that is, whether the attack of the electrophile or the proton expulsion is the rate–determining step.
The answer to all these questions comes from the evidence of isotope effects.
A number of aromatic compounds labeled with deuterium or tritium were subjected to electrophilic aromatic substitution such as bromination, nitration, and Friedel-Crafts alkylation. It was found that in all these reactions the rates of substitution of hydrogen, deuterium and tritium are equal (i.e., deuterium or tritium is replaced at the same rate as protium) indicating that there is more or less no kinetic isotope effect.
If the rates of replacement of the various hydrogen isotopes are the same, it can only imply that the reactions whose rates we are comparing do not involve the breaking of a carbon-hydrogen bond. In other words, the expulsion of the proton does not occur in the rate–determining step.
This interpretation is consistent with the proposed mechanism. The rate of the overall electrophilic aromatic substitution is controlled by the slow bonding of the electrophile to aromatic ring to form the arenium ion (carbocation).
Sulphonation does show a small isotope effect. However, even in sulphonation the overall rate is controlled mainly by step 1.
Thus the absence of isotope effects establishes not only the two-step mechanism of electrophilic aromatic substitution, but also the relative velocities of the steps. Attachment of the electrophile to a carbon atom of the ring is the difficult step (see free-energy diagram) but it is equally difficult whether the carbon carries protium or deuterium or even tritium. The next step, pass of hydrogen ion, is quite easy. Although it occurs more slowly for deuterium (or tritium) than for protium, it really makes no difference. Slightly slower or slightly faster, its speed has no effect on the overall rate. Consider the free-energy diagram more closely:
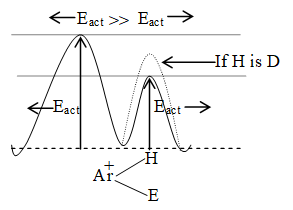
Since the energy barrier to the right (i.e. ahead of the arenium ion) – whether slightly lower for protium or slightly higher for deuterium – is still considerably lower than the barrier to the left (i.e., behind the carbocation), every arenium ion formed, whether I (H) or I (D), goes on to form the product.

The barrier behind the arenium ion is the Eact for the reverse of step 1. It is this reverse reaction that must be much slower than step 2 so that step 1 is essentially the rate–determinining step.
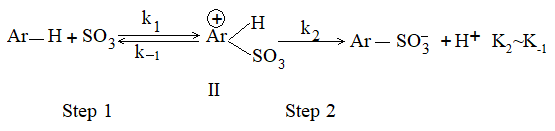
Now one can understand that why nitration and similar reactions are not reversible. In the reverse of nitration, nitrobenzene is protonated (the reverse of step 2) to form the arenium ion I. This is as same as the ion I formed during the nitration process (step 1). Thus, it does the same thing, that is, it reforms nitrobenzene.
Unlike most of the electrophilic aromatic substitution reactions, sulphonation shows a moderate isotope effect, that is, ordinary hydrogen (protium) is displaced form an aromatic ring about twice as fast as deuterium.

However, this does not mean that sulphonation takes place by a different mechanism than nitration, that is one involving a single step or one involving a two-step with step 2 as the rate –determining step.
Let’s consider the rate constants, k, for the various steps:
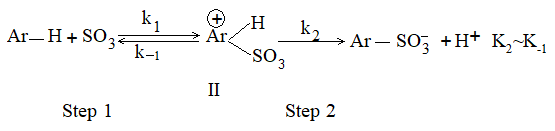
Unlike most of the electrophilic aromatic substitution, sulphonation is reversible. Reversibility implies that arenium ion (II) can lose SO3 to form the hydrocarbon. In sulphonation, step 2 is not much faster than the reverse of step 1. In sulphonation, the energy barriers on either side of the arenium ion are of roughly the same height. Thus some ions can go one way, and others can go the other way. Whether the arenium ion is II (H) or II (D), the barrier to the left (i.e., behind the ion) is of the same height. But to climb the barrier to the right (i.e., ahead the ion), a carbon-hydrogen bond must be broken, thus this barrier is higher for arenium ion II (D) than for arenium ion II (H). Consequently, more deuterated ions than ordinary ions revert to starting material. Thus, overall sulphonation is slower for the deuterated benzene.

The particular shape of potential energy curve that makes sulphonation reversible also permits an isotope effect to be observed.
The absence of the kinetic isotope effect (or primary isotope effect) does not prove the existence of an intermediate, but only gives information about the amount of C-H stretching that occurs in the T.S. Support for this mechanism is afforded by the fact that such intermediates have been isolated. Fior the halogenation of arenes the following conditions favour aromatic halogenation (i.e., nuclear substitution):
a) absence of light,
b) low temperature, and
c) the presence of a halogen carrier ( a Lewis acid)
Benzene does not react with bromine or chlorine unless a Lewis acid is present in the mixture.
Benzene does not decolorize a solution of bromine or chlorine in carbon tetrachloride.
Bromination and chlorination may be very conveniently carried out at ordinary temperature in the presence of a Lewis acid. When Lewis acids are present, benzene reacts readily with bromine or chlorine. The reactions give bromobenzene and chlorobenzene, respectively, in good yields. The Lewis acids most commonly used to bring about bromination and chlorination reactions are the bromides or chlorides of Fe, Al and Sb (FeX3, AlX3 and SbX3), all in the anhydrous form. Ferric bromide and ferric chloride are usually generated in the reaction mixture by adding iron to it. The iron then reacts with halogen to produce the ferric halide:
Thus, iron is most commonly used being converted to the Lewis acid, FeX3, by the halogen.
The mechanism for electrophilic aromatic chlorination is as follows:
Step 1 Chlorine combines with FeCl3 to form the Lewis acid complex that dissociates to form a positive chloride ion and FeCl4–.
Step 2 : The positive chlorine ion (chloronium ion) attacks benzene to form an arenium ion:
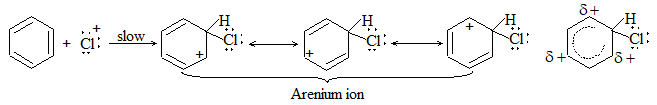
Step 3 : A proton is removed from the arenium ion by FeCl4– to yield chlorobenzene and hydrogen chloride. At the same time, this step regenerates the catalyst, FeCl3.
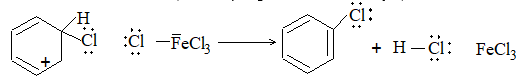
The function of the Lewis acid is apparent in step 1. The ferric chloride reacts with chlorine to yield a positive chlorine ion, Cl+ and a complex FeCl4–. The mechanism of the bromination of benzene in the presence of ferric bromide is analogous to the one for chlorination. Ferric bromide serves the same purpose in aromatic bromination as ferric chloride does in aromatic chlorination, that is, it assists in the generation and transfer of a positive halogen ion.
Aromatic fluorination requires special conditions and special types of apparatus because fluorine reacts much readily with benzene. Moreover, it is difficult to limit the reaction to just monofluorination.
Fluorobenzne can be made by an indirect method.
On the other hand, iodine is so less reactive that some special techniques are to be used to bring about direct iodination. To avoid the reversible reaction, the reaction must be carried out in the presence of an oxidizing agent such as nitric acid.
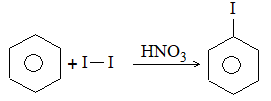
The better method to make iodo-compounds is to use diazonium salts:
Arenediazonium salt
A similar dual mechanism can also operate when halogenation is carried out with hypochlorous or hypobromous acid. This reaction is acid-catalyzed, and the active species could be or positive halogen formed as follows:
Addition of halogens to alkenes similarly involves attack by positive halogen to form an intermediate cation.
Relatively loosely held pi () electrons of an alkene make it more reactive, thus, the positive halogen is transferred from the halogen molecule itself, X2, with loss of X–.
On the other hand, the less reactive benzene molecule needs the assistance of a Lewis acid. The reaction occurs with the loss of the better leaving group, FeCl4–.
More highly reactive aromatic compounds, that is those whose pi () electrons are more available (phenol, aniline) do react with halogens in the absence of any Lewis acid catalyst.
Nitration of arenes
Aromatic nitro-compounds, ArNO2, are usually prepared by direct nitration. For this purpose, any of the following reagents may be employed.
a) Concentrated or fuming nitric acid,
b) Mixed acid (a mixture of concentrated nitric and sulphuric acids,
c) Nitric acid in organic solvents such as acetic acid, nitromethane,
d) Acyl nitrate (acetyl or benzoyl) in organic solvents,
e) Nitronium salts in organic solvents, and
f) Nitrosation followed by oxidation
Of these, the most commonly employed reagent is mixed acid. The others are used according to situations. For example, acetyl nitrate is often useful to get large amounts of the o-nitro-isomer:
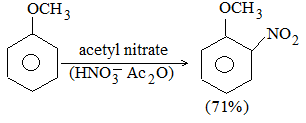
Benzene reacts slowly with hot concentrated nitric acid to yield nitrobenzene. Nitration of benzene becomes much faster if it is carried out by heating benzene with a mixture of concentrated nitric acid and concentrated sulfuric acid.

Concentrated sulphuric acid increases the rate of the reaction by increasing the concentration of the electrophile, the nitronium cation, .
As an outcome of a great deal of experimental work, the following mechanism has been proposed for electrophilic aromatic nitration:
Step 1 In this step the weaker nitric acid accepts a proton from the stronger sulphuric acid:
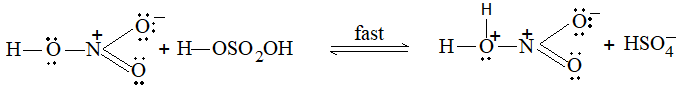
Step 2 Protonated nitric acid dissociates to form a nitronium ion.
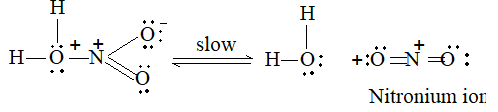
Step 3 Water accepts a proton from another molecule of sulphuric acid:
The overall equation may therefore be written as:
Step 4 Nitronium ion, the actual electrophile in nitration, reacts with benzene to form a resonance stabilized arenium ion:
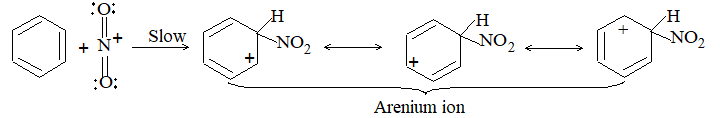
Step 5 A proton is removed by a Lewis base from the arenium ion to yield nitrobenzene:
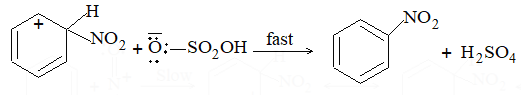
Step (1) generates the nitronium cation, , the electrophile that actually attacks the benzene ring. This reaction is just an acid –base equilibrium in which sulphur acid serves as the acid and the much weaker nitric acid serves as a base.
We may consider that very strong acid, sulphuric acid, causes nitric acid to ionize in the sense HO– …, rather than in the usual way, H+ ….
Evidence for the existence of the nitronium cation comes from
a) Cryoscopic experiments: The van’t Hoff factor, i, is 4. This value satisfies the over all equation given above.
b) Isolation of salts of the nitronium ion : the nitronium ion is well known existing in salts such as nitronium perchlorate (NO2+) (ClO4–), and nitronium tetrafluoroborate (or fluoroborate), (NO2+) (BF4–).
Solutions of these stable nitronium salts in solvents like nitromethane or acetic acid have been known to nitrate aromatic compounds smoothly and in high yield at room temperature.
Sulphonation of benzene
The ease with which aromatic hydrocarbons and their derivatives can be sulphonated, with concentrated or fuming sulphuric acid or with chlorosulphonic acid, is one of their most characteristic properties.
Thus, this reaction can be used to separate alkanes from arenes (aromatic hydrocarbons) because the alkanes are not so readily sulphonated.
Aromatic sulphonic acids are generally prepared by direct sulphonation, since this is far more convenient than indirect methods.
The normally used sulphonating agents are:
a) concentrated sulphuric acid,
b) sulphur trioxide in sulphuric acid, that is, oleum (fuming sulphuric acid),
c) sulphur trioxide in organic solvents such as nitromethane, pyridine, etc,
d) chlorosulphonic acid in carbon tetrachloride (using one molecule of reagent),
e) sulphonyl chloride (using excess of reagent).
Benzene reacts slowly with hot concentrated sulphuric acid to yield benzenesulphonic acid:
But benzene reacts with fuming sulphuric acid at room temperature to produce benzene sulphonic acid:
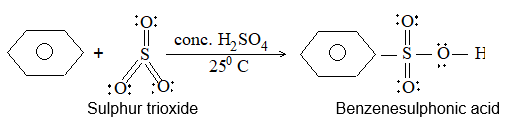
Experimental work based on kinetic studies in concentrated sulphuric acid and in oleum strongly favours the theory that the electrophile is sulphur trioxide.
In concentrated sulphuric acid, sulphur trioxide is produced in an equilibrium in which H2SO4 acts as both an acid and a base.
Sulphonation of many aromatic compounds involves the following steps:
Step 1 Formation of SO3 in concentrated sulphuric acid:
It is simply, an acid-base equilibrium, between molecules of sulphuric acid.
Step 2: Sulphur trioxide, the actual electrophile (as S atom is highly electron deficient due to being attached with three O atoms), attaches itself to the benzene ring to form the intermediate carbocation, an arenium ion :
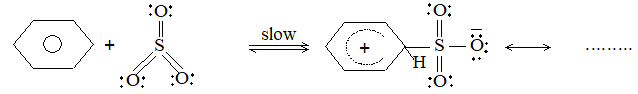
Step 3 The arenium ion loses a proton to a Lewis base to form the benzenesulphonate ion, the anion of benzenesulphonic acid which, being a strong acid is highly ionized:
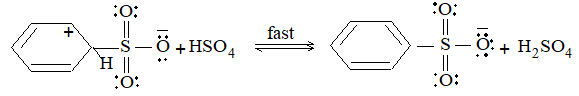
Step 4 A proton is accepted by the benzenesulphonate ion to form benzenesulphonic acid:
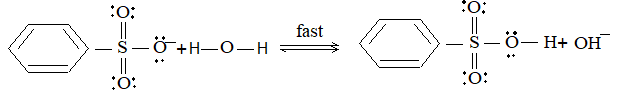
Equilibrium is a far to the left, as benzene sulphonic acid is a strong acid. All of the steps, including step 1 in which sulphur trioxide is formed from sulphuric acid, are in equilibriua. This implies that the overall reaction is also in equilibrium. In concentrated sulphuric acid, the overall equilibrium, the sum of all the steps (1 – 4), is:
In fuming sulphuric acid, containing an excess of SO3, step 1 is meaningless (unimportant) because the dissolved sulfur trioxide reacts directly. Since each of these steps is in equilibrium, the position of equilibrium can be influenced by the experimental conditions we employ:
To sulphonate benzene, we should use concentrated sulphuric acid or better fuming sulphuric acid, so that the position of equilibrium lies appreciably to the right, and benzenesulphonic acid is obtained in good yield.
To remove a sulphonic acid group from a benzene ring we should employ dilute sulphuric acid and pass steam through the mixture, so that with a high concentration of water the equilibrium lies appreciably to the left and desulphonation occurs. The equilibrium is shifted even further to the left with volatile aromatic compounds because they will distil with the steam.
Sulphonation and desulphonation reactions are often used in synthetic work. For instance, we may introduce a sulphonic acid group into a benzene ring to influence the course of some further reaction and later we may remove the sulphonic acid group by desulphonation.
Friedel–Crafts reaction In 1877, Charles Friedel (a French chemist) and James M. Crafts (an American scientist), discovered new methods to prepare alkylbenzenes (ArR) and acylbenzenes (. These reactions are now known as the Friedel–Crafts alkylation (the reaction involving the introduction of an alkyl group into the benzene ring) and the Friedel-Crafts acylation (the reaction involving the introduction of an acyl group into the benzene ring) reactions.
Friedel-Crafts reaction involves the introduction of an alkyl or acyl group into the benzene ring in the presence of a catalyst.
Friedel-Crafts alkylation The following is a general equation for a Friedel-Crafts alkylation reaction:
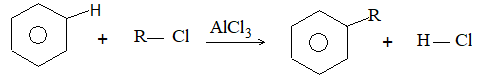
Two possible mechanisms can operate for the Friedel-Crafts alkylation with alkyl halides:
a) One involving an alkyl halide-Lewis acid polar complex (bimolecular mechanism), and
b) The other involving an alkyl carbonium ion (unimolecular mechanism)
In either case, the Friedel-Crafts alkylation can be described as an electrophilic substitution in which the substrate is the aromatic ring.
Just as an acid reacts with a base, similarly an electrophile reacts with a nucleophile, a species that provides the electrons that the electrophile seeks. From the opposite point of view, this reaction can be described as a nucleophilic reaction in which the aromatic ring of the arene is the nucleophile.
It is a well known fact that alkyl halides form 1:1 adducts with Lewis acids, and there is evidence that this adduct reacts with the arene. When R-X is a primary alkyl halide (i.e., the alkyl group is of the straight-chain type), a simple carbocation (probably) does not from. Instead, the aluminium chloride forms a complex with the alkyl halide. This complex acts as the electrophile and reacts with the arene.
In this complex, the carbon-halogen bond is nearly broken and the carbon atom of the alkyl group carries a considerable positive charge:
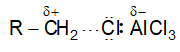
Even though this complex is not a simple carbocation, it acts like a carbocation and thus transfers a positive alkyl group to the aromatic ring.
Theses complexes react so much like carbocations that they also undergo typical carbocation rearrangements.
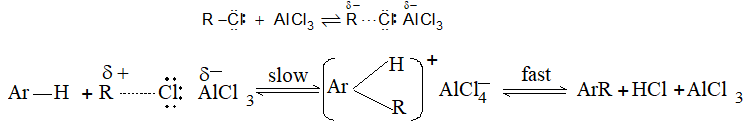
On the other hand, because of the relative stability of s- and t- carbocations, the adducts formed with s- and t- alkyl halides ionize, and form the free carbocation the active species:
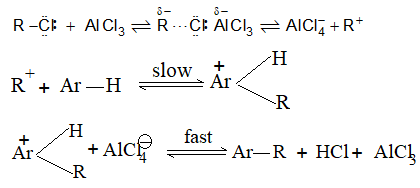
The electrophile is thus either a free carbocation, , or a species like the polar complex that can readily transfer to the aromatic ring. This duality of mechanism is common in electrophilic aromatic substitution.
In either case, the Lewis acid R+ is displaced from RCl by the other Lewis acid, AlCl3.
These mechanisms explain the fact that normal alkyl groups can be fairly introduced without rearrangement at low temperatures as ionization of the adduct is retarded.
They also account for the fact that rearrangement occurs at higher temperatures than those used above. Now, the adduct ionizes to form the free carbocation which rearranges by hydride and/or methyl carbanion shift. For instance, at higher temperatures, n-propyl chloride gives isopropylbenzene:
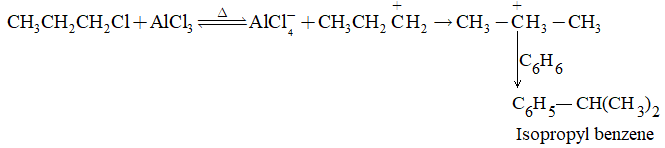
Similarly, we can explain the fact that at higher temperatures, isobutyl chloride gives t-butylbenzene. These two mechanisms are extremes, CH3 – X involved in the adduct and 30 R-X as the carbocation.
The carbocation route is increasingly favoured if the greater is the branching in the alkyl halide. Thus many reactions proceed by a dual mechanism.
Let’s consider the mechanism for the FCAK (Friedel-Crafts Alkylation) with isopropyl chloride as the alkyl halide:
Step 1 It’s a Lewis acid-base reaction between the alkyl halide (Lewis base, L.B) and the aluminium chloride (Lewis acid, L.A.) to form the 1:1 adduct. The complex then dissociates to form a free carbocation and .
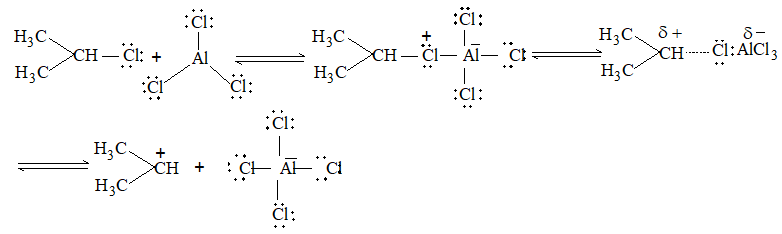
Notice that, the ion is a better leaving group than Cl–. Thus the Lewis acid, AlCl3, serves the same purpose here that a Lowry-Brønsted acid does in the protonation of an alcohol.
Step 2 The free carbocation, behaving as an electrophile, attaches itself to the benzene ring to produce the intermediate carbocation, the arenium ion:
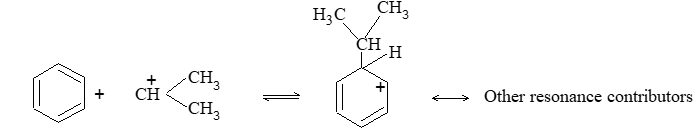
Step 3 The arenium ion loses a proton to a Lewis base to form isopropylbenzene. This step also regenerates the catalyst AlCl3 and liberates HCl.
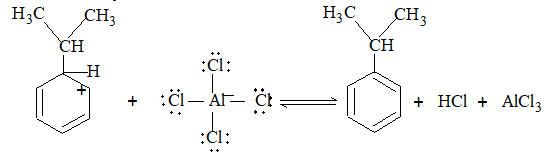
Friedel-Crafts alkylations are not just restricted to the use of alkyl halides and aluminium chloride. Many other pairs of reagents that form the carbocations or polar complexes (species like carbocations) may be used as well.
Thus, the Friedel-Crafts alkylation in its widest sense involves reactants other than alkyl halide and Lewis acids other than aluminium chloride.
The alkylating agents may be
a) alkylhalides,
b) aliphatic alcohols,
c) alkenes,
d) ethers and
e) alkyl esters of organic and inorganic acids.
From the view point of convenience, the alkylating agent is usually confined to alkyl halides, alcohols and alkenes.
Two types of catalysts are used in the Friedel-Crafts reaction.
(i) Lewis acid catalysts Metal halides, their general order of reactivity is :
The more reactive the substrate, the weaker the Lewis acid used.
(ii) Lowry–Brønsted acid catalysts H3PO4, H2SO4 and HF.
Aluminium chloride, originally used by Friedel and Crafts, is the most widely used catalyst. It may be used with any alkylating (or acylating) agent. However, the presence of a small amount of halogen acid is essential, in the case of alkens.
Although aluminium chloride can be used with any alkylating agent, the common practice is to use this catalyst with alkyl halides and alkenes. In other cases we have the following options:
a) Boron trifluoride or hydrogen fluoride with alkenes, and alcohols,
b) Hydrogen fluoride with esters, and
c) Sulphuric acid with alkenes and alcohols.
The ease of alkylation with alkyl halides, using AlCl3 as the catalyst, depends on the nature of the alkyl group and on the halogen atom:
a) For a given halogen atom, the order is
This is also the order for alcohols, esters and ethers.
b) For a given alkyl group, the order is
Alcohols as alkylating agent
A mixture of an alcohol and a lewis acid catalyst may be used:
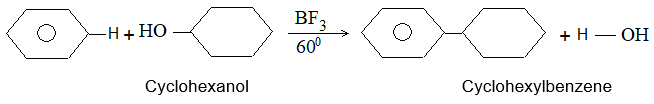
The mechanistic pattern is similar to that of the alkyl halides. However, it appears that, even for a primary alcohol, the carbocation path is followed:
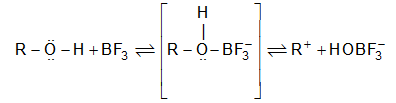
The same mechanism operates for ethers.
Alkenes as alkylating agents A mixture of an alkene and a proton acid catalyst may be used: With proton acid catalysts, the first step is the formation of a carbocation. The carbocation then acts as an electrophile and attacks the aromatic ring, leading to alkylation:
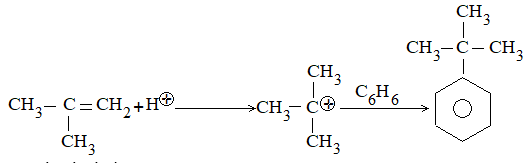
Other examples include:
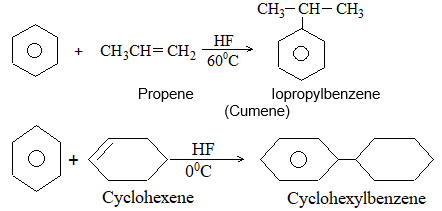
Alkadienes may also be used as alkylating reagent (1, 4-addition):
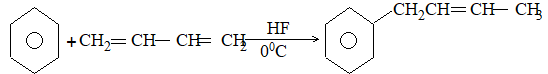
Many kinds of unsaturated compounds other than hydrocarbons may also be used:
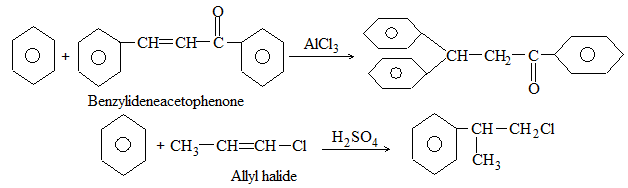
Friedel–Crafts acylation The Friedel-Crafts acylation reaction is an effective method of introducing an aryl group into an aromatic ring.
The group is called an acyl group. A reaction which introduces an acyl group into a compound is called an acylation reaction.
Two common acyl groups are the acetyl group and the benzoyl group:

The benzoyl group, C6H5CO- is not to be confused with the benzyl group, C6H5CH2– .
The Friedel-Crafts acylation, one of the most important modifications of the Friedel-Crafts reaction, is often carried out by treating the aromatic compound with an acyl chloride. Unless the aromatic compound is highly reactive, the reaction requires the addition of at least one equivalent of a Lewis acid, such as AlCl3, as well. An acyl group, RCO- becomes attached to the aromatic ring and the product of the reaction is an acyl ketone, ArCOR.
For example,
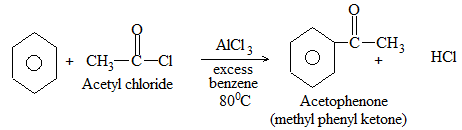
Acyl chlorides, also called acid chlorides, are easily obtained by treating corresponding carboxylic acids with the acid chlorides of inorganic acids:
a) Phosphorus pentachloride, PCl5 (an acid chloride of phosphoric acid)
b) Phosphorus trichloride, PCl3 (an acid chloride of phosphorus acid)
c) Thionyl chloride, SOCl2 (an acid chloride of sulphurous acid)
Besides, acid chlorides, the acylating agents may be carboxylic acid anhydrides, carboxylic acids or esters:

In most Friedel-Crafts acylations the electrophile has to be an acylium ion formed from an acyl halide. Thus, the most likely mechanism for Friedel-Crafts acylation is analogous to the carbocation mechanism for Friedel-Crafts alkylation. It involves the following steps:
Step 1 It is a Lewis acid-base reaction forming the 1 : 1 adduct:
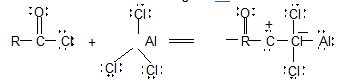
Step 2 The Lewis acid-base complex dissociates to form an acylium ion:
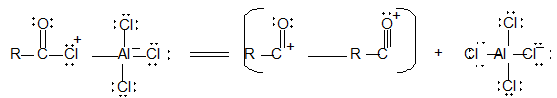
Step 3 The acylium ion, behaving as an electrophile, attaches itself to the benzene ring to form the arenium ion:
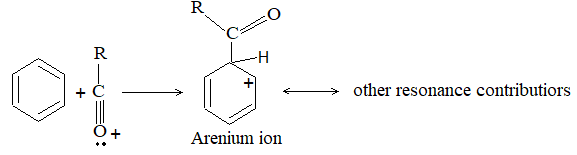
Step 4 : The arenium ion loses a proton to a Lewis base to form the aryl ketone:
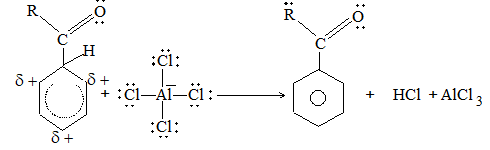
Step 5 The aryl ketone, behaving as a Lewis base, reacts with aluminum chloride (a Lewis acid) to form a complex:
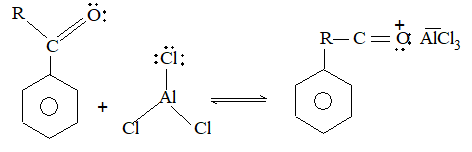
Step 6 Treatment of the complex with water liberates the ketone and hydrolyzes the Lewis acid:
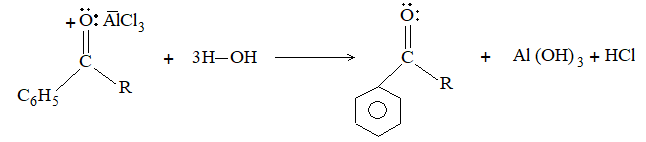
This mechanism fits the pattern of electrophilic aromatic substitution, the attacking reagent, this time, being the acylium ion,
The acylium ion is considerably more stable than ordinary carbocation since in it every atom has an octet of electrons.
There is some evidence that a complex between acid chloride (Lewis base) and aluminum chloride (Lewis acid),
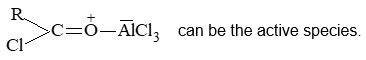
In this case, from the view point of the acid chloride, reaction is acid-catalyzed nucleophilic acyl substitution with the aromatic ring acting as the nucleophile.
As with alkylation, it appears that either mechanism can operate singly or both simultaneously according to circumstances.
Limitations of Friedel–Crafts reactions
The usefulness of Friedel-Crafts reactions is limited by various restrictions:
1) The possibility that the alkyl group will rearrange Friedel-Crafts alkylation is accompanied by a rearrangement that is characteristic of reactions involving carbocations (an evidence in favour of carbocation mechanism).
The structure of the alkyl group plays an important role in the alkylation. For instance, it is easy to introduce a methyl, ethyl or isopropyl group, but usually difficult to introduce n-propyl or n-butyl, since these tend to rearrange.
When the carbocation formed from an alkyl halide, alkene, or alcohol can rearrange to a more stable carbocation (it usually does so, the major product obtained from the reaction is the one from the more stable carbocation.
Isobutyl halides very readily give a t-butyl substitution product:
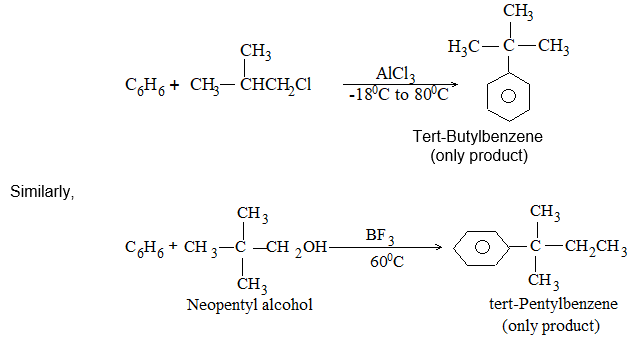
When benzene is alkylated with butyl halide, some of the developing butyl cations rearrange by a hydride shift i.e., some of the developing 10 carbocations become more stable 20 carbocations. Now benzene reacts with both kinds of carbocations to form both n-butylbenzene and sec-butylbenzene:
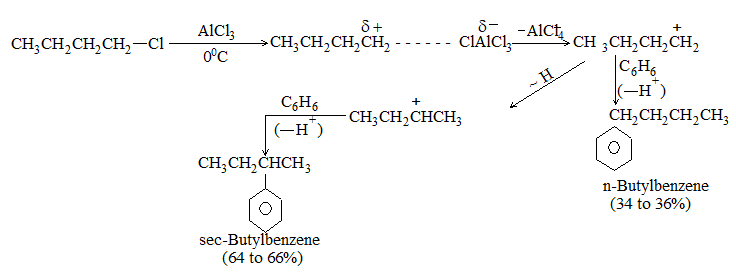
Benzene reacts with n-propyl chloride in the cold, to form n-propylbenzene:
But, isopropylbenzene is the product, if the reaction is carried out at higher temperatures:
At intermediate temperatures, both n-propylbenzene and isoproylbenzene are formed, with latter as the major product:
Using Cyclopropane as the alkylating agent is a very good means of introducing the n-propyl group:
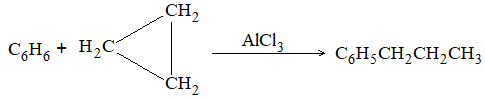
Acid chlorides may be used to introduce long straight-chain groups.
2. The possibility of polysubstitution It is not always possible to stop the reaction at the required stage because there is always a tendency to over-alkylate (particularly with and groups). Alkyl groups are electron-releasing groups. Thus, once an alkyl group is introduced into the benzene ring, it activates the ring towards further substitution:
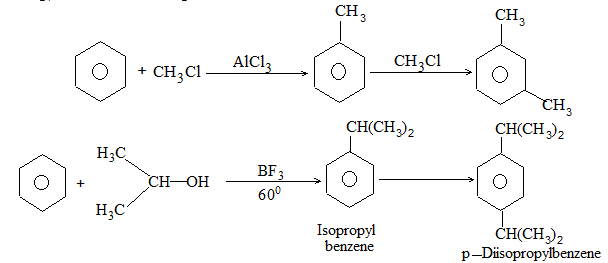
Since the attachment of an alkyl side chain makes the ring more susceptible to further attack, caution must be taken to limit substitution to monoalkylation. This is done by using an excess of the aromatic hydrocarbon (as in halogenation of alkanes) so that the probability of an alkyl carbocation (seeking an aromatic ring) to collide with an unsubstituted ring is more than colliding with a substituted one.
Frequently, the aromatic substrate performs the double duty serving as a solvent as well as a reactant. However, polyacylations are not possible in Friedel-Crafts acylations. The acyl group (RCO-) is an electron-withdrawing group. When it forms a complex with AlCl3 in the last step of the reaction, it becomes even more electron withdrawing. This strongly prohibits further substitution making monoacylation easy.
3) Unreactivity of aryl and vinyl halides Aryl halides and vinylic halides can’t be used as alkylating agents the because of the low reactivity of halogen attached to an aromatic ring and to a doubly bonded carbon, respectively. Moreover, they do not form carbocations readily.
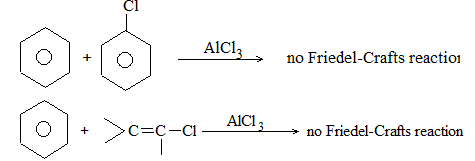
4) An aromatic ring less reactive than that of the halobenzene does not undergo the Friedel–Crafts reaction
When powerful electron-withdrawing groups are present on the aromatic ring, the Friedel-Crafts reactions usually give poor yields. This applies to both alkylations and acylations.
Groups (substituents) present on an aromatic ring can have a large effect on the reactivity of the ring towards electrophilic aromatic substitution.
Electron withdrawing substituents make the ring less reactive by making it electron deficient. Any substituent more electron withdrawing (or deactivating) than a halogen, that is, any meta-directing group, makes an aromatic ring too electron-deficient to undergo a Friedel-Crafts reaction. Therefore, the following compounds usually give poor yields in Friedel-Crafts reactions:
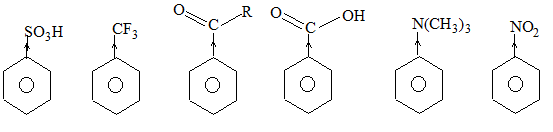
5) Aromatic rings containing the –NH2, –NHR or –NR2 group do not undergo Friedel–Crafts reaction When the aromatic ring bears an -NH2, -NHR, or -NR2 group, the Friedel-Crafts reactions usually give poor yields. This applies to both alkylations and acylations. The strongly basic nitrogen ties up the Lewis acid needed for ionization of the alkyl halide. Thus, the amino groups -NH2, -NHR, and -NR2 are changed into powerful electron–withdrawing groups by the Lewis acids needed to catalyze Friedel-Crafts reactions.
For example,
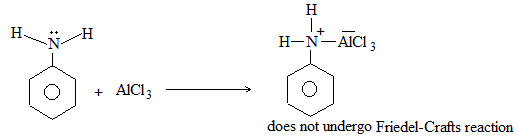
6) Rearrangement of a group In the Friedel-Crafts reaction, an alkyl group may be removed from one molecule and transferred to another (disproportionation) or may migrate from one position to another in the ring.
In spite of these limitations, the Friedel-Crafts reaction in its various modifications is extremely useful synthetic tool.
Synthetic applications of Friedel–Crafts acylation A straight-chain alkyl group longer than ethyl generally cannot be attached in good yield to an aromatic ring by Friedel-Crafts alkylation because of the rearrangement of the carbocation.
Such a group is readily introduced in two steps:
(1) formation of ketone by Friedel-Crafts acylation
(2) Clemmensen or Wolff-Kishner reduction of the ketone.
Rearrangements of the carbon chain do not occur in Friedel-Crafts acylations. The acylium ion is more stable than most other carbocations because it is stabilized by resonance. Hence, there is no driving force for a rearrangement.
Because rearrangements do not occur, Friedel-Crafts acylations followed by reduction of the carbonyl group to a CH2 group often give us much better routes to unbranched alkylbenzenes than do Friedel-Crafts alkylations.
Suppose we want to synthesize n-propyl benzene. If we try to synthesize this through a Friedel-Crafts alkylation, a rearrangement takes place and the major product is isopropylbenzene:
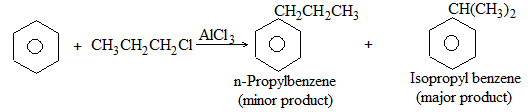
On the other hand, the Friedel-Crafts acylation of benzene with propanoyl chloride yields a ketone without any rearrangement of the carbon chain in an excellent yield.
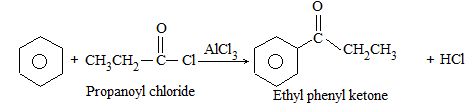
Now, this ketone can be reduced to propylbenzene by several methods. One of these general methods is called the Clemmensen reduction. It consists of refluxing the ketone with hydrochloric acid containing amalgamated zinc:
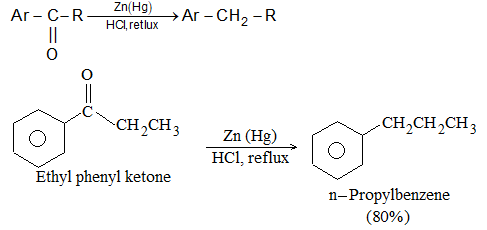
Friedel-Crafts acylation followed by ketone reduction is the synthetic equivalent of Friedel-Crafts alkylation.
Zinc and hydrochloric acid also reduce nitro groups to amino groups.
Other examples:
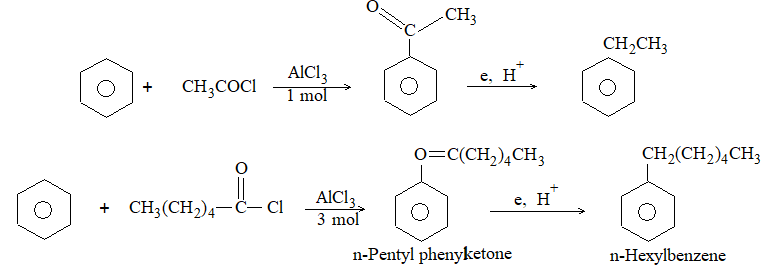
Another general method of reduction-called the Wolff-Kishner reduction – consists of heating the ketone with hydrazine and a base:
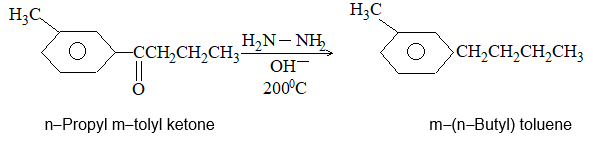
The Friedel-Crafts arylation can provide an effective method to add a new ring to an aromatic compound if cyclic anhydrides are used as acylating agents:

Exercise 6: (i) Benzene gives mainly
(A) substitution reaction
(B) addition reaction
(C) elimination reaction
(D) all of these
(ii) The compound that is most reactive towards electrophilic substitution is
(A) toluene
(B) benzene
(C) nitrobenzene
(D) benzoic acid
(iii) Which of the following is meta-directing group?
(A) –COOH
(B) –OH
(C) –NH2
(D) – Cl
Exercise 7: (i) The function of anhydrous aluminium chloride in the Friedel-Crafts reaction is
(A) to absorb water
(B) to absorb hydrochloric acid
(C) to produce an electrophile
(D) to produce nucleophile
(ii) Substitution reactions of benzene are mainly
(A) free radical type
(B) electrophilic type
(C) nucleophilic type
(D) None of these
(iii) In the nitration of benzene with a mixture of conc. HNO3 and conc. H2SO4, the active species involved is
(A) NO3
(B) NO2
(C) NO2–
(D) NO2+
Effect of substituents on reactivity and orientation
Only one monosubstituted compound is obtained, when one substituent group is introduced into the benzene ring. However, when a second group is introduced, three isomeric disubstituted benzenes are possible: ortho-, meta- and para-.
Through experimental investigation it was found that when the second substituent enters the benzene ring, the main product is
i) either a mixture of the o- and p- isomers
ii) or the m-
Pure o-, p- or m- substitution is rare, as all the three possible isomers are obtained simultaneously. Since the rates at which they are formed are very different, the slowest one results in the formation of very little of that derivative.
Experimental investigations also showed that usually o, p-substitution is associated with activation of the benzene ring, i.e., reaction is faster than in benzene itself, while m-substitution is associated with deactivation of the ring, i.e., reaction is slower than in benzene.
Thus, substituent groups already present on the ring affect both the rate of the reaction and the site of attack, when substituted benzenes undergo electrophilic attack.
In other words, we can say that substituent groups affect both the reactivity of the ring and the orientation of substitution in electrophilic aromatic substitution, that is, when an electrophile attacks an aromatic ring, it is the group already attached to the ring that determines how readily the attack occurs and where it occurs.
According to their influence on the reactivity of the ring, we can divide substituent groups into two classes:
i) Activating groups, those that make the ring to be more reactive than benzene itself
ii) Deactivating groups, those that make the ring to be less reactive than benzene itself
Also, according to the way they influence the orientation of attack by the incoming electrophile, we can divide substituent groups into two classes.
i) ortho, para directors: Substituents in this class tend to bring about electrophilic substitution chiefly at the positions ortho and para to themselves.
These groups are called ortho–para directors because they tend to direct the incoming group primarily into the ortho and para positions.
ii) meta directors: Substituents in this category tend to bring about electrophilic substitution chiefly at the position meta to them.
These groups are called meta directors because they cause attack to occur mainly at meta position. Like benzene, toluene undergoes electrophilic aromatic substitution, for example, sulphonation. Although, three monosulphonation products are possible, this reaction actually yields considerable amounts of only two of them-the ortho and para isomers:
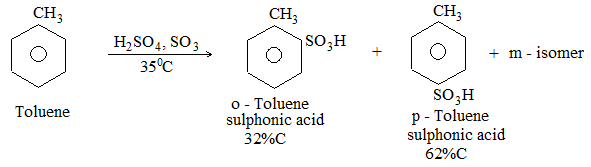
Both benzene and toluene are insoluble in sulphuric acid, while the sulphonic acids are readily soluble. Thus, completion of reaction is indicated simply by disappearance of the hydrocarbon layer. Benzene reacts completely within 20 to 30 minutes while toluene reacts within only a minute or two, when they are shaken with fuming sulphuric acid at room temperature.
Similar results are obtained when toluene is subjected to nitration, halogenation and Friedel-Crafts reactions. When we nitrate with nitric and sulphuric acids, we get mononitrotoluenes in the following relative amounts:
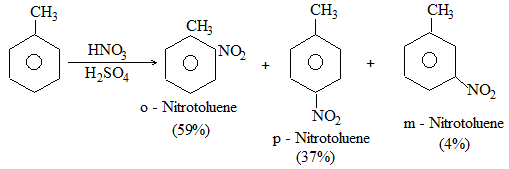
Of the mononitrotoluenes obtained from the reaction, 96% (59% + 37%) have the nitro group in an ortho or para position, Only 4% have the nitro group in a meta position.
This implies that, in some way, the methyl group makes the ring more reactive than unsubstituted benzene and directs the attacking reagent to the ortho and para positions of the ring.
All alkyl groups are activating groups, and also they are all ortho, para directors.
The methoxyl group, CH3O- and the acetamido group, CH3CONH-, are strong activating groups, and both are ortho para directors.
The hydroxyl group and the amino group are very powerful activating groups and are also powerful ortho-para directors.
Both phenol and aniline react with bromine in water, in the absence of a catalyst, to produce products in which both of the ortho positions and the para position are substituted. These tribromo products are obtained in nearly quantitative yield:
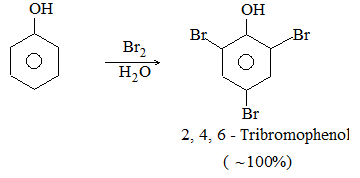
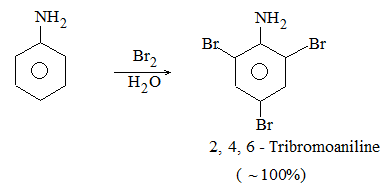
On the other hand, the nitro group is a very strong deactivating group. Nitrobenzene undergoes nitration at a rate only 1/10,000 times that of benzene.
The nitro group is a meta director; when nitrobenzene is nitrated with nitric acid and sulphuric acid, 93% of the substitution occurs at the meta position:
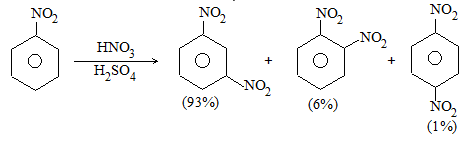
Similarly, the carboxyl group (- CO2H), the sulpho group (- SO3H) and the trifluoromethyl group (-CF3) are also deactivating and meta directors.
How to determine orientation
It is quite simple to determine the effect of a group on orientation. It involves the following steps:
1) The compound containing the substituent group whose orientation is to be investigated is allowed to undergo substitution.
2) The resulting product is analyzed for the relative proportions of the three isomers.
3) The identification of each isomer as ortho, meta or para through comparison with an authentic sample of that isomer, prepared by some other method from a compound of known structure.
This way it has been found that every substituent group can be placed into one of two classes:
i) ortho, para-directors
ii) meta directors
Of the five positions open to attack, three (60%) are ortho and para to the substituent group, and two (40%) are meta to the group.
If there were no selectivity in the substitution reaction we would expect the ortho and para isomers to make up 60% of the product and the meta isomer to make up 40%. When nitration is carried out in a number of substituted benzenes, we observe that groups such as -OH, -NHCOCH3, -CH3, -F, -Cl, -Br, and -I direct 96-100% of nitration to the ortho and para positions while the groups of the type -NO2, , -CN, -COOH, -SO3H and -CHO direct 72-94% of nitration to the meta positions.
Further investigation revealed that a given group causes the same general kind of orientation either predominantly ortho, para or predominantly meta – whatever the electrophile involved.
However, the actual distribution of isomers varies from reaction to reaction.
How to determine relative reactivity
A substituent group is labeled as activating if the benzene ring carrying the group is more reactive than benzene. If the ring carrying the group is less reactive than benzene it is classified as deactivating.
The reactivities of benzene and the substituted benzene can be compared in the following ways:
i) Reactions are carried out under identical conditions and the time–needed for their completion is measured.
For example, toluene reacts with fuming sulphuric acid in about one-tenth to one-twentieth time than required by benzene. This indicates that, toluene is more reactive than benzene, and therefore, -CH3 is an activating group.
ii) Reactions are completed within the same period of time and the severity of experimental conditions is observed.
For example, benzene is nitrated in less than an hour at 600C by a mixture of concentrated nitric acid and concentrated sulphuric acid while comparable nitration of nitrobenzene requires treatment at 900C with fuming nitric acid and concentrated sulphuric acid. Evidently, nitrobenzene is less reactive than benzene, and the nitro group, -NO2, is a deactivating group.
Classification of substituents
Almost all substituent groups fall into one of two classes:
Class I makes the benzene ring relatively more reactive and directs the incoming group mainly to the o- and p- positions, that is, activating and ortho-para directors:
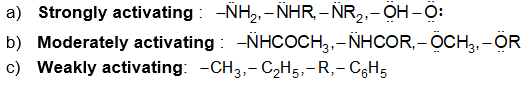
Class II makes the benzene ring relatively less reactive and directs the incoming group mainly to the m-position, that is, deactivating and meta-directing:
a) Strongly deactivating:
b) Moderately deactivating :
Halo substituents are weakly deactivating but ortho-para directors:
![]()
Now, one can predict fairly accurately the course of hundreds of aromatic substitution reactions, just by knowing the results summarized above. For instance, one can now predict that bromination of nitrobenzene will yield mainly the meta isomer and that the reaction will go more slowly than the bromination of benzene itself. Actually, it requires severe conditions to go at all.
Similarly, we can predict that nitration of acetanilide (C6H5 NHCOCH3) will yield mainly the ortho and para-isomers, and will occur more rapidly than nitration of benzene.
Although it is possible to account for these effects in a reasonable way, it is essential to memorize the above classification, so that you may deal rapidly with synthetic problems involving aromatic compounds.
Orientation in disubstituted benzenes
The problem of orientation gets complicated by the presence of two substituents on the ring. However, one can frequently make very definite predictions. The position occupied by a third group entering the ring depends on the nature of the two groups already present.
Experimental investigations have shown that:
(i) When both the substituent groups belong to class I, the directive power of each group is decided by the following order:
The more powerful activating group generally determines the outcome of the reaction.
For instance, one can consider the orientation of electrophilic substitution of p-methylacetanilide: The acetamide group is a much stronger activator than the methyl group. Thus, the acetamide group determines the outcome of the reaction, and hence substitution occurs mainly at the position ortho to the acetamido group:
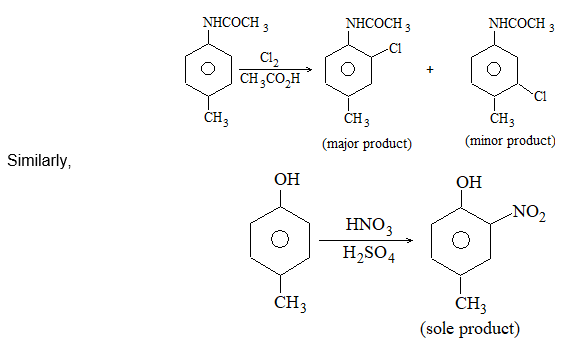
In the following examples, the number of arrow heads indicates (qualitatively) the amount of substitution, and the number below indicates the number of isomers:
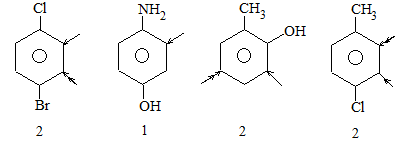
ii) When both the substituent groups belong to class II, then it is difficult to introduce a third group. The directive influence (power) of each group is usually the following:
iii) When the two groups direct the incoming electrophile differently, i.e., belong to classes I and II, then class I takes precedence i.e., the ortho-para director determines the orientation of the incoming group because all ortho-para directors are more activators than meta directing, deactivators.
The following example illustrates the fact that strongly activating groups generally win out over deactivating groups:
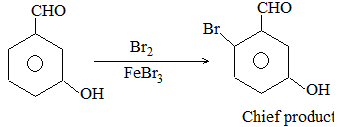
iv) Steric effects also play a crucial role in aromatic substitutions. For instance, substitution does not take place to an appreciable extent between meta substituents (i.e., the ortho to both the groups) if another position is vacant as illustrated by the nitration of m-bromochlorobenzene:
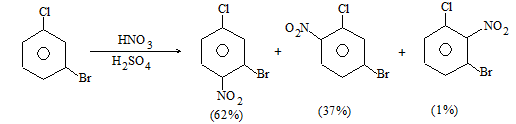
Notice that only 1% of the mono-nitro product has the nitro group between the bromine and chlorine.
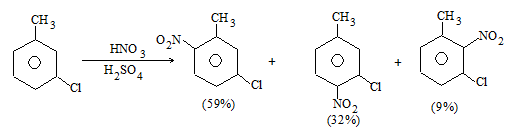
And here only 9% of the mono-nitro product has the nitro group between the methyl and chlorine. Both these examples clearly indicate that there is often little substitution between two groups that are meta to each other.
v) When the two substituents are located such that the directive influence of one reinforces that of the other, the third group enters almost completely at the reinforced position. Thus in the following examples, the orientation clearly must be that indicated by the arrows:
When the directive influence of one group opposes that of the other, it may be difficult to predict the major product. In such cases complicated mixtures of several produces are often obtained.
There must be a fairly large difference in the effects of the two groups for clear-cut results, otherwise one gets results of the following kind:
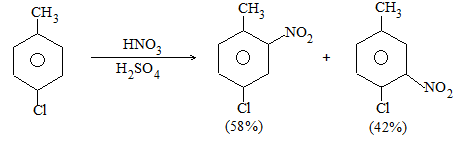
Theory of substituent effects
Certain groups activate the benzene ring toward electrophilic substitution and direct incoming group to ortho and para positions, whereas other groups deactivate the ring and (except halogens) direct substitution to meta positions.
Reactivity and orientation are both matter of relative rates of reaction. When we say that a group (such as -NH2) activates the ring we mean that the group increases the relative rate of the reaction. Thus, an aromatic compound with an activating group reacts faster (in electrophilic substitutions) than benzene, similarly the activator causes ortho, para orientation because it makes the ortho and para positions react faster than the meta positions.
When we say that a group deactivates the ring, we mean that an aromatic compound with a deactivator reacts slower than benzene, similarly the deactivator causes meta orientation because it makes the meta positions react faster than the ortho, para positions.
Irrespective of the reagent involved, the rate of electrophilic aromatic substitution is determined by the same slow step i.e., the attack of the electrophile on the ring to form the carbocation called arenium ion:
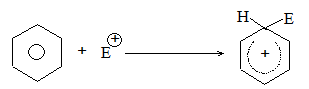
Since this is the rate -determining step, any differences in rate of substitution must be due to differences in the rate of this step.
For closely related reactions (such as electrophilic aromatic substitutions), a difference in rate of formation of intermediate carbocations is mainly determined by a difference in Eact, i.e., by a difference in stability of transition states.
Thus, we can account for relative reaction rates by examining the transition state for the rate – controlling steps.
Any factor that increases the energy of the transition state relative to that of the reactants decreases the relative rate of the reaction because, of the increase of the free energy of activation of the reaction. Similarly, any factor that decreases the energy of the transition state relative to that of the reactants lowers the free energy of activation and hence increases the relative rate of the reaction.
Let’s focus on the rate – determining step of electrophilic substitution of substituted benzenes that is the step resulting in the formation of the arenium ion. By using the letter G to represent any ring substituent including hydrogen (If G is hydrogen, the compound is benzene itself), we can write the formula for a substituted benzene in a generalized way:
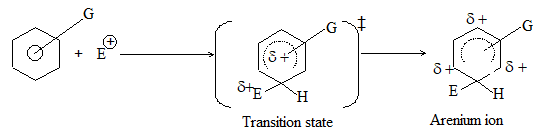
We can also write the generalized structure for the arenium ion in the similar manner. By this representation we mean that G can be in any position – ortho, meta, or para – relative to the electrophile, E. Using these conventions, we are able to write the rate -determining step for electrophilic aromatic substitutions in the general way.
Examination of the rate controlling step (RCS) for a large number of reactions indicates that the relative rates of the reactions depend on whether G releases or withdraws electrons. If G is an electron-releasing group (relative to hydrogen), the reaction occurs faster than the corresponding reaction of benzene:
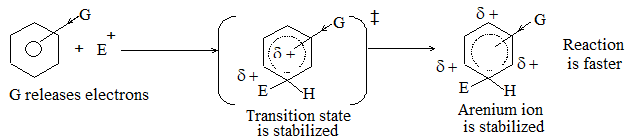
If G is an electron-withdrawing group, the reaction is slower than that of benzene:
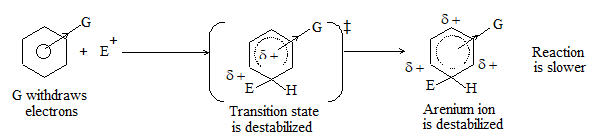
This happens because the substituent (G) affects the stability of the transition state relative to that of the reactants.
Electron-releasing groups apparently make the transition state more stable, whereas electron-withdrawing groups make it less stable. This is understandable, because the transition state resembles the arenium ion, and the arenium ion is a delocalized carbocation.
Factors that stabilize the intermediate carbocation by dispersing the positive charge also stabilize (for the same reaction) the incipient carbocation of the transition state.
The arenium ion is high-energy intermediate and the step that leads to it is a highly endothermic step. Thus, (according to the Hammond-Leffler postulate) there should be a strong resemblance between the arenium ion itself and the transition state.
Since the arenium ion is positively charged, an electron-releasing group stabilizes it (by dispersing the positive charge) and also the transition state leading to the arenium ion, because the transition state is a developing delocalized carbocation.
Similar kind of arguments is valid about the effect of electron-withdrawing groups. An electron-withdrawing group makes the arenium ion less stable, and correspondingly makes the transition state leading to the arenium ion less stable.
The following figure shows clearly, how the electron-withdrawing and electron-releasing abilities of substituents affect the relative free energies of activation of electrophilic aromatic substitution reactions.
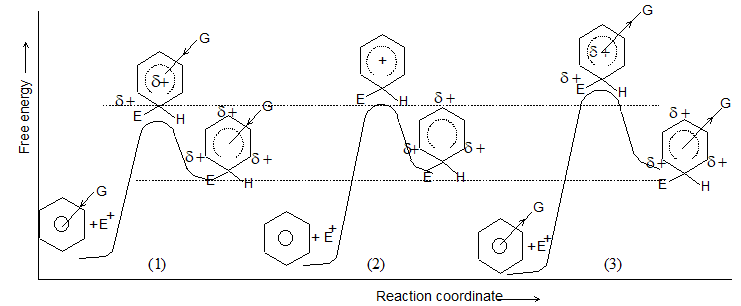
The above figure shows a comparison of free-energy profiles for arenium ion formation in a ring with an electron-releasing substituent (G), no substituent, and an electron-withdrawing substituent (→G). In (1), an electron-donating group (G), stabilizes the transition state. The energy of activation barrier is lowest, and therefore the reaction is the fastest. In (3) the electron-withdrawing group (→G), raises the transition state energy by destabilizing it. The energy of activation barrier is the highest, and therefore the reaction is the slowest. Middle energy profile (2) with no substituent serves as a reference for comparison.
In electrophilic aromatic substitution the intermediate carbocation (arenium ion) is a hybrid of three structures (I, II and III) in which the positive charge is distributed about the ring, being strongest at the ortho and para positions to the carbon atom attacked by the electrophile:
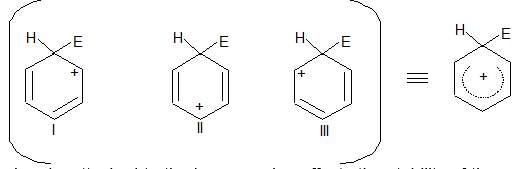
A group already, attached to the benzene ring affects the stability of the carbocation by dispersing or intensifying the positive charge, depending upon its electron-releasing or electron withdrawing nature. It is clear from the structure of the ion (I to III) that this stabilizing or destabilizing effect is especially important when the group is attached ortho or para to the carbon being attacked.
Factors determining orientation in aromatic substitution reactions
We can explain the electron-releasing and electron-withdrawing properties of substituent groups on the basis of two factors:
i) Inductive effects and
ii) Resonance effects
These two factors also determine orientation in electrophilic aromatic substitution reactions.
To account for rates of substitution in benzene, toluene and nitrobenzene, we can compare the structures of the carbocations formed from these compounds:
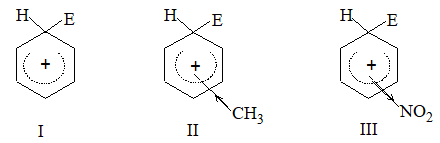
Methyl group has +ve inductive effect. By releasing electrons the methyl group tends to nullity the positive charge in the ring. In this process methyl group becomes positive itself. This dispersal (or delocalization) of the charge stabilizes the carbocation.
Similarly the +ve inductive effect of the methyl group disperses the developing positive charge in the transition state and thus leads to a faster reaction.
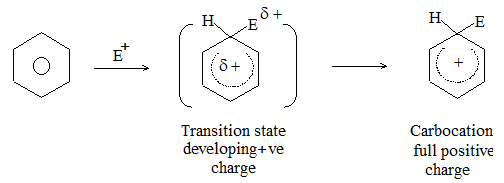
On the other hand the -NO2 group has -ve inductive effect. By withdrawing electrons, the nitro group tends to intensify the positive charge. This destabilizes the carbocation, and thus leads to a slower reaction. Like -CH3, other alkyl groups also have +ve inductive effect. Thus they release electrons, and like -CH3 they activate the ring. For example, the reactivity of tert-butylbenzene is 16-17 times greater than that of benzene, towards nitration. All the halogens are more electronegative than carbon and thus exert an electron-withdrawing inductive effect.
In other groups having an electron-withdrawing inductive effect the atom directly attached to the ring carries either partial or full positive charge. Look at the following examples:
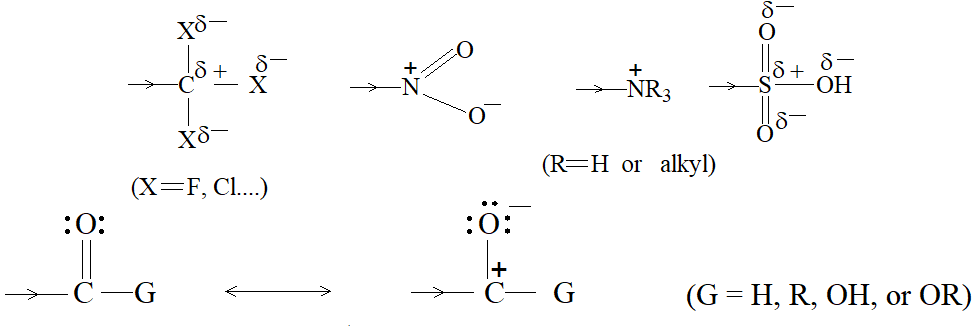
The full-fledged positive charge of the group has powerful attraction for electrons. In the other deactivating groups (such as -NO2, -CN, -COOH) the atom next to the ring is attached by a multiple bond to oxygen or nitrogen. These electronegative atoms attract the mobile electrons, making the atom next to the ring electron-deficient. To make up this deficiency, the atom next to the benzene ring withdraws electrons from the ring.
Replacement of hydrogen in -CH3 by halogen decreases the electron-releasing, tendency of the group, and ultimately converts it into an electron-withdrawing group for higher substituted alkyl groups.
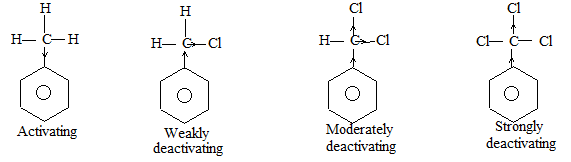
Toward nitration, toluene is 25 times as reactive as benzene, benzyl chloride is only one third as reactive as benzene. The -CH2Cl group is thus weakly deactivating. Further replacement of hydrogen by halogen yields the -CHCl2 and the -CCl3 groups and results in stronger deactivation. The trifluoromethyl group, because of the three highly electronegative fluorine atoms, is strongly electron withdrawing. It is a strong deactivating group and a powerful meta director in electrophilic aromatic substitution reactions.
The trifluoromethyl group affects the rate of reaction by making the transition state (leading to the arenium ion) to be highly unstable. It does this destabilization by withdrawing electrons from the developing carbocation. This increases the positive charge on the ring.
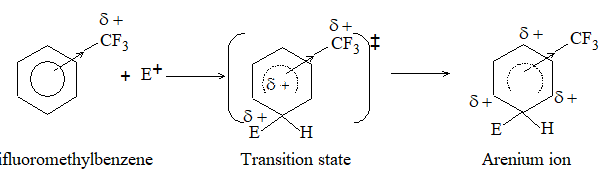
An activating group activates all positions of the benzene ring. Even the positions meta to it are more reactive than any single position in benzene itself. It directs ortho and para simply because it activates the ortho and para positions much more than it does the meta. A deactivating group deactivates all position in the ring, even the positions meta to it. It directs meta simply because it deactivates the ortho and para positions even more than it does the meta. Thus both ortho, para orientation and meta orientation arise in the same way: the effect of any group – whether activating or deactivating – is strongest at the ortho and para positions.
Let us compare the carbocations formed by attack at the ortho and meta positions of toluene (a compound that contains an activating group). Each of these is a hybrid of three structures, I-III for ortho and IV – VI for meta.
In one of these three structures, III, the positive charge is residing on the carbon atom to which -CH3 is attached. Although -CH3 releases electrons to all positions of the ring, it does so most strongly to carbon atom nearest to it. Consequently, structure III is especially more stable.
Ortho attack
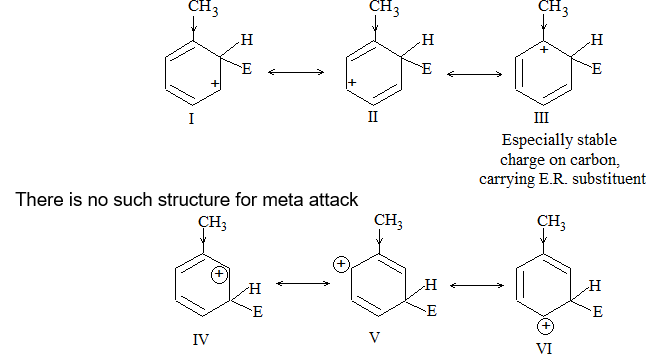
Because of contribution from structure III, the hybrid carbocation resulting from attack at the ortho position is more stable than the carbocation resulting from attack at a meta position. Therefore, ortho substitution occurs faster than meta substitution.
Similarly, it can be seen that attack at the para position (VII – IX) also yields a more stable carbocation, through contribution from VIII than attack at the meta position:
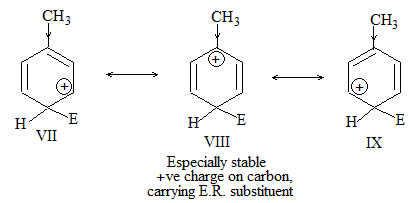
Thus, in toluene, ortho, para substitution is faster than meta substitution because electron release by -CH3 is more effective during attack at the positions ortho and para to it.
Next, we can compare the carbocations formed by attack at the ortho and meta positions of nitrobenzene (a compound that carries a deactivating group).
Each of these is a hybrid of three structures, I-III for ortho attack, IV -VI for meta attack.
Ortho attack
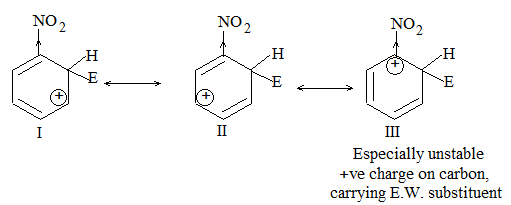
In one of the three structures, III, the positive charge is residing on the carbon atom to which -NO2 is attached. Although NO2 withdraws electrons from all positions, it does so most from the carbon atom nearest to it. Hence, this carbon atom, already positive, has little tendency to accommodate the positive charge of the carbocation. Thus structure III is especially unstable one and does little to help stabilize the ion resulting from attack at the ortho position.
The ion for ortho attack is practically (virtually) a hybrid of only two structures, I and II. Thus, the positive charge is mainly restricted to only two carbon atoms. It is less stable than the ion resulting from attack at a meta position, which is practically a hybrid of three structures. In these contributors the positive charge is accommodated by three carbon atoms.
More the resonating structures of an intermediate or structure, more is the stability.
Meta attack
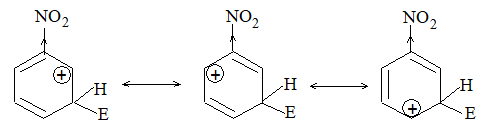
Therefore, ortho substitution occurs more slowly than meta substitution.
In the same way it can be seen that attack at a para position (VII – IX) yields a less stable carbocation, because of the instability of VIII, than attack at a meta position.
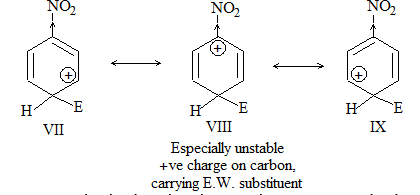
Thus, in nitrobenzene, ortho, para substitution is slower than meta substitution because electron withdrawal by -NO2 is more effective during attack at the positions ortho and para to it.
Both ortho, para orientation by activating groups and meta orientation by deactivating groups follow logically from the structure of the intermediate carbocation.
The charge of the carbocation is always strongest at the positions which are ortho and para to the point of attack. Hence a group attached to one of these positions exerts the strongest effect irrespective of its nature (whether activating or deactivating).
By examining the resonance structures for the arenium ion that would be formed when an electrophile attacks the ortho, meta and para positions of trifluoromethyl methyl benzenes, we can understand how the trifluoro methyl group affects orientation in electrophilic aromatic substitution.
Ortho attack
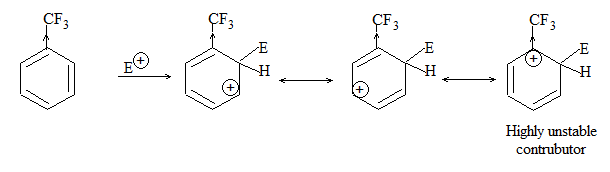
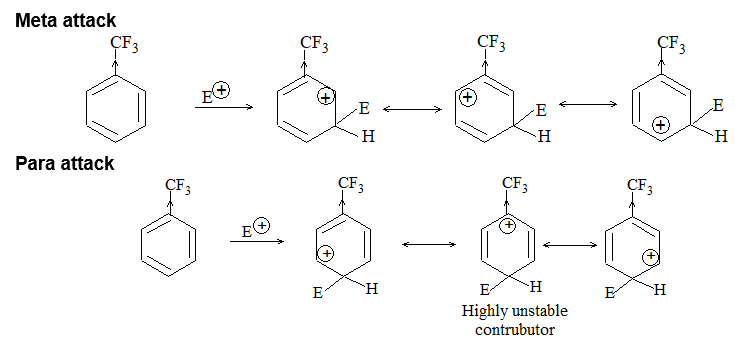
Investigation shows that, in the resonance structures for the arenium ion arising from ortho and para attack, one of the contributing structures is highly unstable relative to all the others because the positive charge is located on the ring carbon bearing the electron withdrawing group. No such highly unstable resonance structure for the arenium ion arises from meta attack.
This implies that the arenium ion formed by meta attack is the most stable of the three. Consequently the transition state leading to the meta substituted arenium ion is the most stable and, therefore, meta attack is favored.
This is supported by experiments which show that the trifluoromethyl group is a powerful meta director:
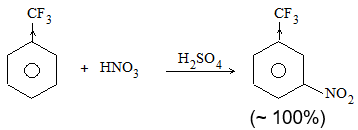
Meta substitution is favoured only in the sense that it is the least unfavorable of three unfavorable pathways. The free energy of activation for substitution at the meta position of trifluoromethylbenzene is less than that for attack at an ortho or para position but it is still far greater than that for an attack on benzene. In other words substitution at the meta position of trifluoromethylbenzene occurs faster than substitution occurring at the ortho and para positions, but it occurs much more slowly than it does with benzene.
All meta-directing groups have either a partial positive charge or a full positive charge on the atom directly attached to the ring.
The nitro group, the carboxyl group, and other meta directing groups are all powerful electron-withdrawing groups and act in a similar way.
The inductive effect of a substituent G arises from the electrostatic interaction of the polarized bond to G with the developing positive charge in the ring as it is attacked by an electrophile. If G is more electronegative atom (or group) than carbon, then the ring will be at the positive end of the dipole:

Attack by an electrophile will be retarded because this will lead to an additional full positive charge on the ring.
Resonance effect
A substituent group affects both the reactivity and orientation in electrophilic aromatic substitution by its tendency to release or withdraw electrons. Electron release and electron withdrawal through inductive effects arise due to the electronegativity of the group concerned.
But certain groups such as -NH2 and -OH, and their derivatives act as powerful activators toward electrophilic aromatic substitution, even though they contain electronegative atoms having electron-withdrawing inductive effects.
These groups release electrons by a resonance effect. The nitrogen of the -NH2 group, in spite of the high electronegativity, is basic and tends to share its lone pair of electrons and acquire a positive charge. Just as ammonia accepts a proton to form the ammonium ion (NH4+), the organic compounds related to ammonia accept protons to form substituted ammonium ions.
The -OH group shows similar but weaker basicity:
The effects of -NH2 and -OH on electrophilic substitution can be accounted for by considering that N and O can share more than a pair of electrons with the ring to accommodate a positive charge.
When G is an atom bearing one or more nonbonding electron pairs, it may lend extra stability to the arenium ion by providing a fourth resonance contributor in which the +ve charge resides on G:
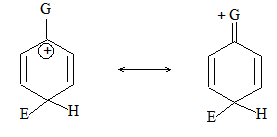
The following order is the application of the electron-donating resonance effect with decreasing order:
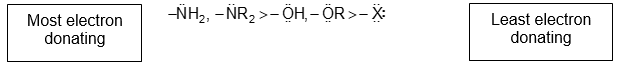
The order of the activating ability of these groups is also the same. Amino groups are highly activating, hydroxyl and alkoxyl groups are somewhat less activating and halogen substituents are weakly deactivating.
When X = F this order can be explained on the basis of the electronegativity of the atoms with the nonbonding pair. The more electronegative, the atom is, the less able it is to accept the positive charge. Of the elements mentioned above, fluorine is the most electronegative and nitrogen the least. When X = Cl, Br, or I, the relatively poor electron donating ability of the halogens (by resonance) can be understood on a different basis.
These atoms (Cl, Br, and I) are all larger than carbon. Therefore, the orbitals that contain the nonbonding pairs are further from the nucleus. Consequently, they do not overlap well with the 2p orbital of carbon.
In general, resonance effects are not transmitted well between atoms of different rows in the periodic table.
Except for the alkyl and phenyl substituents, all of the ortho – para directing groups have at least one pair of nonbonding electrons on the atom adjacent to the benzene ring:
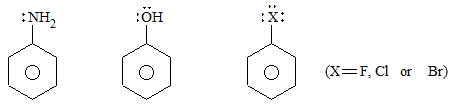
This structural feature i.e., the presence of an unshared electron pair on the atom adjacent to the ring determines the orientation and influences reactivity in electrophilic substitution reactions.
The directive effect of these groups with an unshared pair is mainly due to an electron-releasing effect. It operates mainly in the arenium ion and consequently in the transition state leading to it. To understand these resonance effects, we discuss the effect of the amino group on electrophilic aromatic substitution reactions.
The amino group is not only a powerful activating group but also a powerful ortho-para director. For example, aniline reacts with bromine in aqueous solution at room temperature and in the absence of a catalyst to yield a product in which both ortho positions and the para position are substituted.
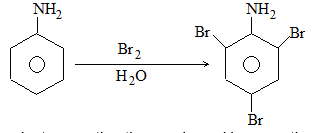
Nitrogen is more electronegative than carbon. However the difference between the electronegativities of nitrogen and carbon in aniline is not large because the carbon of the benzene ring is sp2 hybridized. Thus it is somewhat more electronegative than it would be if it were sp3 hybridized. In electrophilic aromatic substitution, the resonance effect of the amino group is far more important than its inductive effect. Thus, this resonance effect makes the amino group electron releasing.
By writing the resonance structures for the arenium ions that would arise from ortho, meta and para attack on aniline, one can understand this resonance effect.
Ortho attack
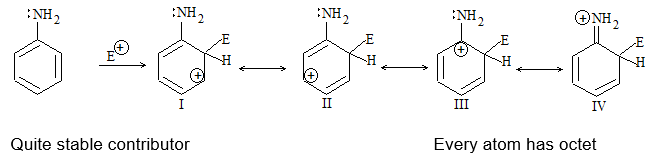
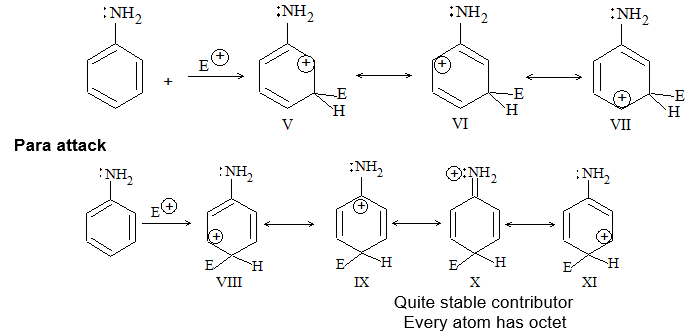
One can see that four reasonable resonance structures can be written for the arenium ions formed by attack at ortho and para positions to the -NH2 group of aniline, whereas only three can be written for the arenium ion obtained from meta attack. This implies that the ortho- and para- substituted arenium ions are more stable.
Of the four contributors for the ortho and para attack, the three structures carry positive charge on the carbons of the ring while the fourth one carries the positive charge on nitrogen.
The relatively stable structures contributing to the hybrid for the ortho- and para- substituted arenium ions play an important role.
In these structures, nonbonding pairs of electrons from nitrogen form an extra bond to the carbon of the ring. Moreover, every atom (except hydrogen) in these structures has a complete octet of electrons. This extra bond and the fact that every atom in each of these structures has a complete outer octet of electrons, makes these structures the most stable of all the contributors.
Because these structures are unusually stable, they make a large and stabilizing contribution to the hybrid.
Therefore, the ortho- and para- substituted arenium ions are considerably more stable than the arenium ion resulting from the meta attack. As a result, the transition states leading to the ortho- and para -substituted ions occur at unusually low free energies. Consequently, electrophiles react at the ortho and para positions very rapidly.
The carbocation (IV to X) is much more stable than the one obtained by attack on benzene itself, or the one obtained (V – VII) from attack meta to the -NH2 group of aniline.
In neither of these, cases, a structure like (IV or X) is possible.
Similarly we can understand that, is more stable than . Here also, it is not a matter of, which atom, nitrogen or carbon, can better accommodate a positive charge; it is a matter of which atom has a complete octet of electrons.
In exactly the same way activation and ortho, para orientation by the -OH group can be explained through the contribution of structures like:
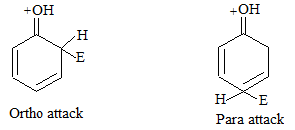
The similar effects of the derivatives of -NH2 and -OH groups are explained with the help of similar structures, shown for para attack only:
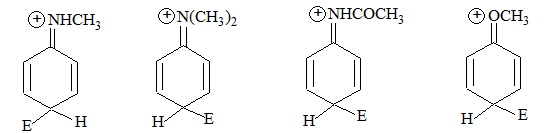
There are two ways of studying the relative reactivities of positions in substituted benzenes:
i) Charge distribution
ii) Stability of the intermediate carbocation.
Orientation based on charge distribution
The charge distribution in monosubstituted benzene depends on the polar effect of the group (i.e., the inductive and / or resonance effect). When the group present in the ring is -OH, -OR, -NH2, etc., the product of further substitution is mainly ortho, and para.
A common feature of all these groups is that the atom adjacent to the nucleus i.e., the key atom carries at least one nonbonding (lone) pair of electrons.
The resonance effect due to this pair of electrons gives rise to increased electron densities in the ortho & para positions:
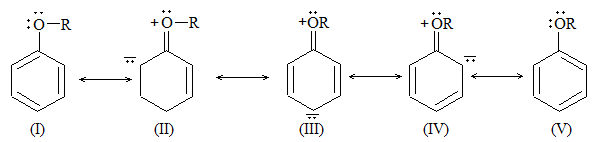
(I) and (V) are the normal structures
(II) and (IV) are o-quinonoid structures (ortho-quinonoid)
(III) is p – quinonoid structure (para-quinonoid)
*Quinonoid means quinine like, since these structure resemble quinine,
![]()
One can see that C6H5OR, is a resonance hybrid of the five resonance structures (I – V).
Thus, the actual state of C6H5OR is a molecule which carries small negative charges at the two o-positions and at the p- position. As a result, substitution with electrophilic reagents takes place in these positions.
Owing to the increased electron-density in the o, p-positions, the substituent group (-OR) present in the ring is associated with activation of the ring. In other words, the presence of an o, p- orienting group facilitates further substitution.
Inductive effect is a type of electron displacement along a carbon chain in which one terminal carbon is joined to an atom (or group) other than H.
The product of further substitution is mainly meta (m-), when the group present in the ring is NO2, CO2H, COR, SO3H, etc.
All these groups have at least one strongly electron attracting atom and a double or triple bond conjugated to the benzene ring.
Thus, they cause an electron displacement away from the benzene ring (nucleus) and towards the group (i.e., a -R effect).
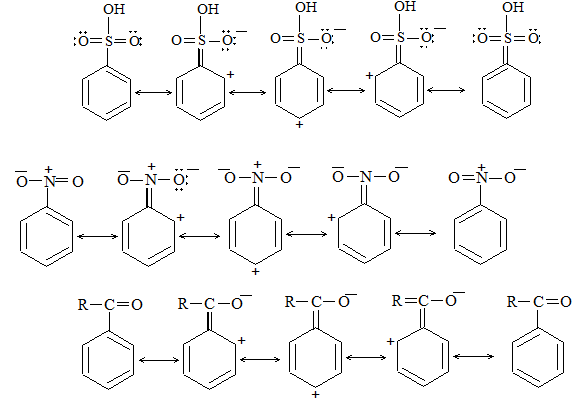
One can see that each of the three compounds C6H5SO3H, C6H5NO2 and C6H5COR is a resonance hybrid of five resonating structures.
Thus, the actual states of C6H5SO3H, C6H5NO2 and C6H5COR are molecules which have small positive charges in the o- and p- positions.
This implies that the m-positions have a relatively high electron density with respect to the o, p-positions. Therefore, these groups are m-orienting to electrophilic reagents.
It also means that for these groups the activation energy for m-substitution is lower than that for o, p-substitution.
Notice that, the relative high electron density of the m-positions with respect to the o, p-positions is due to the withdrawal of electrons from the o, p-positions (i.e., -R effect) rather than a gain of electrons in the m-positions.
Since all m-orienting groups contain at least one strongly electron-attracting atom, the inductive effect of this atom also helps in the withdrawal of electrons from the o, p-positions.
Hence, the presence of these substituents is associated with deactivation of the ring i.e., further substitution is made more difficult by the presence of a m-orienting group.
The directive influence and reactivity effects of halo-substituents
These two effects of halo substituents seem to be contradictory as the halo groups are the only ortho-para directors that are deactivating. All other deactivating groups are meta directors.
We can easily explain the unusual behavior of halo substituents the moment we realize the fact that their electron-withdrawing inductive effect influences reactivity and their electron-donating resonance effect governs orientation.
Let’s consider chlorobenzene. Since the chlorine atom is highly electronegative, it withdraws electrons from the benzene ring and deactivates it:
When a electrophile attacks, the chlorine atom stabilizes the arenium ions formed from the attack at the ortho and para positions relative to that resulting from meta attack.
The chlorine atom does this by donating an unshared pair of electrons. These electrons give rise to relatively stable resonance structures contributing to the hybrids for the ortho-and para substituted arenium ions:
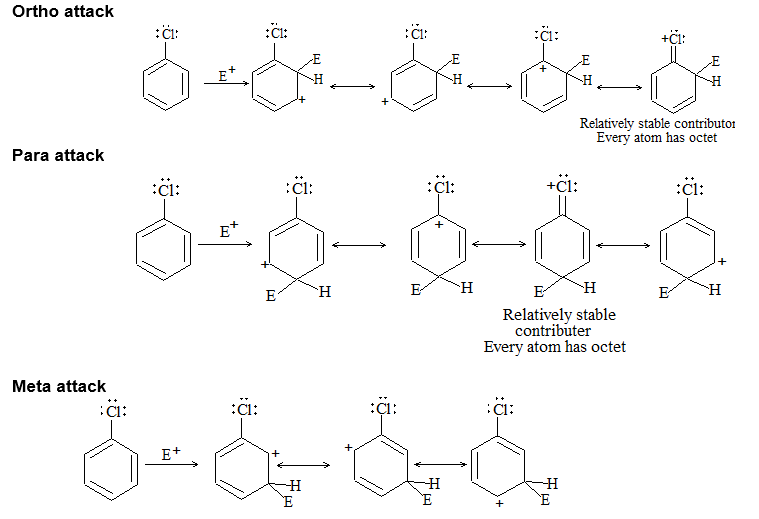
The same thing is valid for bromobenzene.
Summary of the inductive and resonance effects of halo substituents Halo substituents make the ring more electron deficient than that of benzene, through their electron-withdrawing inductive effect. As a result the free energy of activation for any electrophilic aromatic substitution reaction becomes greater than that for benzene itself. Therefore, halo substituents are deactivating. However, halo substituents cause the free energies of activation leading to ortho and para substitution to be lower than the free energy of activation leading to meta substitution, through their electron-donating resonance effect. This makes halo substituents ortho-para directors.
Chlorine has -I (negative inductive effect) and +R (positive resonance effect) since the direction of the C-Cl dipole is in the CCl direction, it follows that the -I effect is greater than the + R effect.
That the +R effect operates is supported by the fact that the chlorine atom is less negative then expected (compared with RCl).
Orientation and reactivity of alkylbenzenes
Alkyl groups are better electron-releasing groups than hydrogen. Because of this, they can stabilize the transition state leading to the arenium ion. This activates the benzene ring toward electrophilic aromatic substitution:
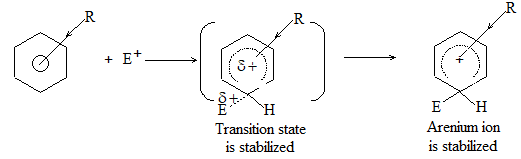
As a result, the free energy of activation of the step leading to the arenium ion, is lower for an alkylbenzene than that for benzene. Consequently alkylbenzenes react faster.
Alkyl groups are ortho-para directions. We can account for this behavior of alkyl groups by writing resonance structures for the arenium ions formed when toluene undergoes electrophilic substitution.
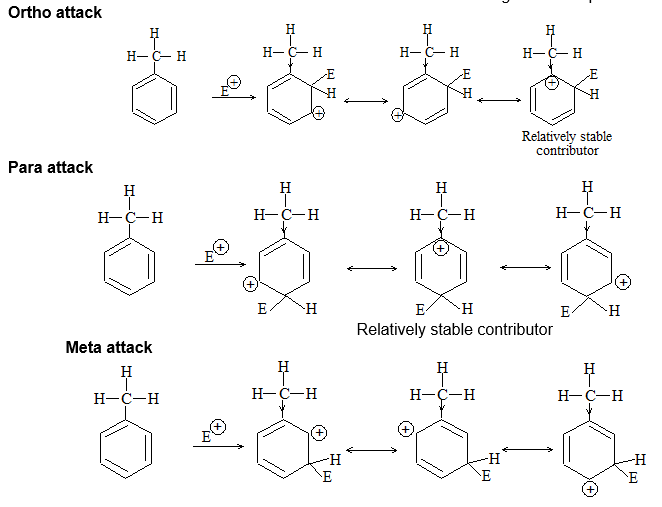
One can see that, for ortho and para attack, we can write resonance structures in which the methyl group is directly attached to a positively charged carbon of the ring. These structures are more stable in comparison to any of the others because in them the stabilizing effect of the methyl group (by electron release) is most effective. Therefore, these structures make a large (stabilizing) contribution to the overall hybrid for ortho and para substituted arenium ions. No such relatively stable structure contributes to the hybrid for the meta-substituted arenium ion. As a result it is less stable than the ortho – or para substituted arenium ion.
Since the ortho- and para – substituted arenium ions are more stable, the transition states leading to them appear at lower energy. Hence, ortho and para substitutions take place most rapidly. Since the order of the inductive effect of alkyl groups is:
methyl < ethyl < propyl < isopropyl < t–butyl
The activating effect of an alkyl group (if entirely due to the + I effect) would also be in the same order. Actually in a number of cases the order is the reverse.
Baker–Nathan effect: The general order of inductive effect of alkyl groups is:
methyl < ethyl < propyl < isopropyl < t-butyl
This inductive order satisfactorily explains various physical data. However, in some cases the inductive order is reversed. The reaction between p-substituted benzyl bromides and pyridine was examined kinetically (Baker and Nathan). The reaction was carried out in acetone. It was sound to be entirely SN2.
Thus, the reaction may be formulated as:
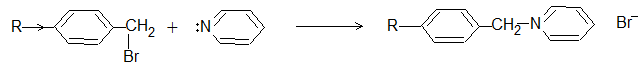
The greater the + I, effect of the R group, the faster the reaction should be. But it was found that the rate order for the alkyl group was:
Methyl > ethyl > isopropyl > t-butyl
This implies that the order of electron release is just the reverse of the inductive effect order. This further indicates that alkyl groups must possess some different mechanism of electron-release in which the order is:
Further investigation showed that in all cases where the usual inductive effect order of alkyl groups was reversed, the alkyl group was attached to an aromatic nucleus. This is led Baker and Nathan to suggest that whenever the H-C bond is attached to an unsaturated carbon atom, the s-electrons of the H-C bond get delocalized (i.e. become less localized) by entering into partial conjugation with the attached unsaturated system. This is definitely the , -conjugation:
This type of conjugation between electrons of single and those of multiple bonds is known as hyperconjugation. It is a permanent name. This name was given by Mulliken, 1941.
A widely used way of looking at the hyperconjugative effect is, to regard the H-C bond as possessing partial ionic character due to resonance.
For example, propene may be written as a resonance hybrid as follows:
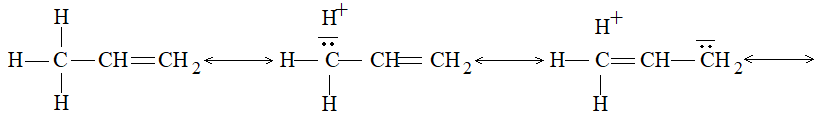
From this view point, hyperconjugation may be regarded as ‘no bond resonance’. The hydrogen atoms are not free. The hyperconjugative effect increases the ionic character of the C-H bond, so that the electrons become partially delocalized through conjugation. Hyperconjugation is greatest in the methyl group and least in the t-butyl group.
Hyperconjugation (along with the -I effect) explains the m-orienting power of the CCl3 group.
Exercise 8: (i) Disubstituted derivatives of benzene are of …….. type/types
(A) 1 (B) 2 (C) 3 (D) 6
(ii) In the halogenation of aromatic nucleus, the halogen carrier, is used to generate the species
(A) Cl– (B) Cl+ (C) Cl– (D) Cl
Exercise 9: (i) The reaction of chlorine with ioluene in presence of ferric chloride gives predominantly
(A) benzoyl chloride
(B) m-chlorotoluene
(C) benzyl chloride
(D) o- and p-chlorotoluene
(ii) Toluene may be oxidized to benzaldehyde by the use of
(A) KMnO4 + H2SO4
(B) K2Cr2O7 + H2SO4
(C) CrO2Cl2
(D) All of these
Answer to Exercises
Exercise 1: (i) D (ii) B (iii) C
Exercise 2: (i) C (ii) C (iii) B
Exercise 3: (i) C (ii) C (iii) C
Exercise 4: (i) A (ii) C (iii) D
Exercise 5: (i) A (ii) C (iii) D
Exercise 6: (i) A (ii) A (iii) A
Exercise 7: (i) C (ii) B (iii) D
Exercise 8: (i) C (ii) B
Exercise 9: (i) D (ii) C
FORMULAE AND CONCEPTS AT A GLANCE
1. Hydrocarbons are broadly classified into
(a) open chain hydrocarbons (acyclic)
(b) closed chain hydrocarbons(cyclic)
2. Open chain hydrocarbons or aliphatic hydrocarbons are further divided into
(a) saturated hydrocarbons – Alkanes
(b) unsaturated hydrocarbons – (i) Alkenes, (ii) Alkynes
3. Alkanes are the simplest organic compounds, also known as paraffins. Each carbon atom here is sp3 hybridized.
4. Alkanes can be prepared by catalytic hydrogenation of unsaturated hydrocarbons, by the reduction of alkyl halides, by the reduction of alcohols, aldehydes, ketones and fatty acids, by condensing two molecules of alkyl halides.
5. Other preparation of alkanes includes
(a) Frankland reaction,
(b) Kolbe’s electrolysis
6. Alkenes are characterized by the presence of a double bond between two carbon atoms. The hybridization of carbon here is sp2.
7. Alkenes can be prepared by dehydration of alcohols, by the dehydrohalogenation of alkyl halides, by the dehalogenation of vicinal dihalides and Kolbe’s electrolysis method.
8. Alkynes are characterized by the presence of a triple bond between two carbon atoms. Each carbon here is sp hybridized.
9. Alkynes can be prepared by dehydrohalogenation of vicinal dihalides, by dehalogenation of vicinal tetrahalides, by Kolbe,s electrolysis method and by hydrolysis of calcium carbide.
SOLVED PROBLEMS
1.
C2 is rotated anticlockwise 120° about C2 – C3 bond. The resulting conformer is
(A) partially eclipsed
(B) eclipsed
(C) gauche
(D) staggered
Sol. (C).

The resulting conformer is a gauche conformer.
2.
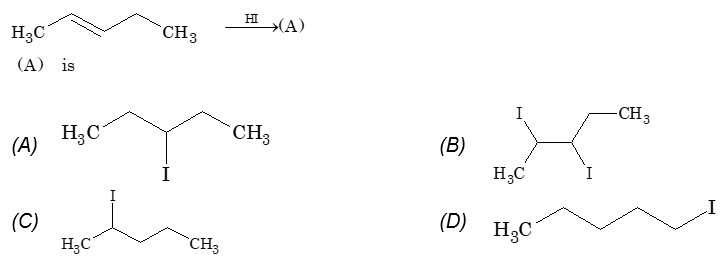
Sol. (C).
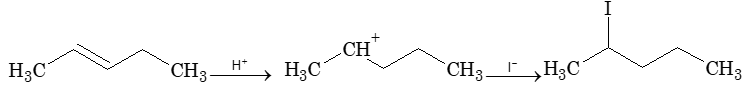
3. How many chiral compounds are possible on mono chlorination of 2-methyl butane?
(A) 2 (B) 4 (C) 6 (D) 8
Sol. (A).
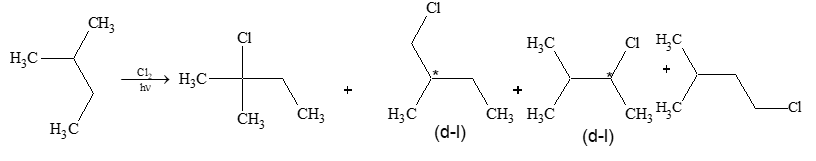
4. Which of the following represents the given mode of hybridization sp2 – sp2 – sp – sp from left to right?

Sol.
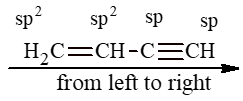
5. Identify the reagent from the following list which can easily distinguish between 1-butyne and 2-butyne.
(A) bromine, CCl4
(B) H2, Lindlar catalyst
(C) dilute H2SO4, HgSO4
(D) ammoniacal Cu2Cl2 solution
Sol. (D). 2-butyne will not react with ammoniacal Cu2Cl2.
6.
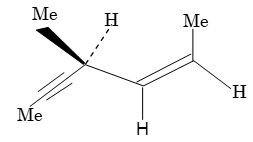
Hydrogenation of the above compound in the presence of poisoned palladium catalyst gives
(A) an optically active compound
(B) an optically inactive compound
(C) a racemic mixture
(D) a diastereomeric mixture
Sol. (B). Due to the formation of the plane of symmetry by the syn-addition of hydrogen in the triple bond.
7. In the presence of peroxide, hydrogen chloride and hydrogen iodide do not give anti-Markonikov’s addition to alkenes because
(A) both are highly ionic
(B) one is oxidizing and the other is reducing
(C) one of the steps is endothermic in both the cases
(D) all the steps are exothermic in both the cases
Sol. (C). Since, I– is reducing agent, but Cl– is not oxidizing, but one of the steps is endothermic in both cases.
8. Propyne and propene can be distinguished by
(A) conc. H2SO4
(B) Br2 in CCl4
(C) dil. KMnO4
(D) AgNO3 in ammonia
Sol. (D). Propyne is having active hydrogen.
9. Ethylene on treatment with CH3COCl gives
(A) 4 chlorobutan-2-one
(B) 4 chloro butan-1-one
(C) 3 cholorbutan-2-one
(D) 3 chlorobutan-1-one
Sol. (A).
10. Ethylene on treatment with H2SO4, followed by hydrolysis gives,
(A) methyl alcohol
(B) ethyl alcohol
(C) propyl alcohol
(D) none of them
Sol. (B).